Arsenic
L'arsenic est l'élément chimique de numéro atomique 33, noté par le symbole As. Son corps simple se présente sous la forme d'un solide cristallin argenté.
| Arsenic | |||||||||||
 Échantillon d'arsenic. | |||||||||||
| |||||||||||
| Position dans le tableau périodique | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Symbole | As | ||||||||||
| Nom | Arsenic | ||||||||||
| Numéro atomique | 33 | ||||||||||
| Groupe | 15 | ||||||||||
| Période | 4e période | ||||||||||
| Bloc | Bloc p | ||||||||||
| Famille d'éléments | Métalloïde | ||||||||||
| Configuration électronique | [Ar] 3d10 4s2 4p3 | ||||||||||
| Électrons par niveau d’énergie | 2, 8, 18, 5 | ||||||||||
| Propriétés atomiques de l'élément | |||||||||||
| Masse atomique | 74,921 595 ± 0,000 006 u | ||||||||||
| Rayon atomique (calc) | 115 pm (114 pm) | ||||||||||
| Rayon de covalence | 119 ± 4 pm[1] | ||||||||||
| Rayon de van der Waals | 185 pm | ||||||||||
| État d’oxydation | ±3, 5 | ||||||||||
| Électronégativité (Pauling) | 2,18 | ||||||||||
| Oxyde | Acide faible | ||||||||||
| Énergies d’ionisation[2] | |||||||||||
| 1re : 9,788 6 eV | 2e : 18,589 2 eV | ||||||||||
| 3e : 28,351 eV | 4e : 50,13 eV | ||||||||||
| 5e : 62,63 eV | 6e : 127,6 eV | ||||||||||
| Isotopes les plus stables | |||||||||||
| Propriétés physiques du corps simple | |||||||||||
| État ordinaire | Solide | ||||||||||
| Allotrope à l'état standard | Arsenic gris (rhomboédrique) | ||||||||||
| Autres allotropes | Arsenic jaune (cubique centré), arsenic noir (orthorhombique) | ||||||||||
| Masse volumique | 5,72 g·cm-3 (gris); 1,97 g·cm-3 (jaune); 4,7–5,1 g·cm-3 (noir)[3] |
||||||||||
| Système cristallin | Rhomboédrique | ||||||||||
| Dureté (Mohs) | 3,5 | ||||||||||
| Couleur | gris métallique | ||||||||||
| Point de fusion | 817 °C (36 bar), pas de fusion à la pression normale[3] |
||||||||||
| Point d’ébullition | 613 °C (sublimation)[3] | ||||||||||
| Énergie de fusion | 369,9 kJ·mol-1 | ||||||||||
| Énergie de vaporisation | 34,76 kJ·mol-1 | ||||||||||
| Volume molaire | 12,95×10-6 m3·mol-1 | ||||||||||
| Pression de vapeur | 7,5×10-3 mmHg (280 °C); 7,5×10-2 mmHg (323 °C); |
||||||||||
| Chaleur massique | 330 J·kg-1·K-1 | ||||||||||
| Conductivité électrique | 3,45×106 S·m-1 | ||||||||||
| Conductivité thermique | 50 W·m-1·K-1 | ||||||||||
| Divers | |||||||||||
| No CAS | [5] | ||||||||||
| No ECHA | 100.028.316 | ||||||||||
| No CE | 231-148-6 | ||||||||||
| Précautions | |||||||||||
| SGH[3] | |||||||||||
  Danger |
|||||||||||
| SIMDUT[6] | |||||||||||
 D1A, D2A, |
|||||||||||
| Transport[3] | |||||||||||
| Classification du CIRC | |||||||||||
| Groupe 1 : Cancérogène pour l'homme[7] | |||||||||||
| Unités du SI & CNTP, sauf indication contraire. | |||||||||||

L'arsenic appartient au groupe des pnictogènes (groupe 15) avec l'azote (N), le phosphore (P), l'antimoine (Sb), le bismuth (Bi) et le moscovium (Mc). Il a des propriétés intermédiaires entre celles des métaux et des non-métaux, comme l'antimoine dont il est proche. Il est généralement considéré comme un métalloïde. C'est un élément hautement toxique et un polluant réglementé depuis 2005 en Europe par une directive[8].
Le groupe des pnictogènes montre une tendance croissante à former des sulfures stables plutôt que des oxydes. De même, les ions à base d'As, Sb et Bi sont précipités par le sulfure d'hydrogène en solution.
L’arsenic est chimiquement très semblable au phosphore, élément non-métal qui le précède dans le même groupe. On dit qu'il est son « analogue chimique ». Il présente aussi une grande analogie avec l'antimoine semi-métallique, plus lourd, qui le suit dans le groupe.
Cette matière connue depuis l'Antiquité est aussi un perturbateur endocrinien[9].
Isotopes
L'arsenic possède 33 isotopes connus, de nombre de masse variant de 60 à 92, ainsi qu'au moins 10 isomères nucléaires. Seul un de ces isotopes, 75As, est stable, faisant donc de l'arsenic un élément monoisotopique. Cet isotope étant également le seul présent dans la nature, l'arsenic est donc également un élément mononucléidique. Sa masse atomique est de 74,921 60(2) u.
Les radioisotopes de l'arsenic les plus stables sont 73As, qui a une demi-vie de 80 jours, suivi de 74As (17,7 jours) et 76As (1 jour). 78As a une demi-vie de 90 minutes, mais tous les autres isotopes ont une demi-vie inférieure à 1 heure et la plupart inférieure à 1 minute.
Occurrences dans les milieux naturels, géologie et minéralogie
L'arsenic corps simple polymorphe existe à l'état natif au moins sous deux variétés allotropiques, qui sont autant d'espèces minérales définies de la catégorie élément natif, soient l'arsenic natif et l'arsénolamprite. De grandes masses d'arsenic natif, accumulées lentement par l'effet des eaux acides d'infiltrations minières, ont été trouvées dans les anciennes mines de Sainte-Marie-aux-Mines.

Il existe assez communément dans la nature des arséniures et sulfo-arséniures de fer, de nickel ou de cobalt, qui attestent des combinaisons faciles de l'élément arsenic avec le soufre et nombre de métaux. L'arsenic est souvent associé à l'antimoine, comme le prouve le stibarsen. Il est en particulier assez souvent associé aux métaux précieux comme l'or et l'argent.
L'arsenic est un élément perturbateur occasionnellement retrouvé dans les gisements aurifères. Il est considéré comme un ennemi par les métallurgistes. Plusieurs compagnies minières le fuient, mais les plus courageuses et tenaces en sont récompensées. L'emploi de main-d'œuvre extrêmement compétente peut compenser l'apport négatif de cet élément. Une expertise dans l'extraction profonde de tellures aux côtés de géologues de haut niveau s'avère un atout non négligeable pour la compagnie exploitant le gisement.

Il est également présent, sous diverses formes ioniques ou de composés de combinaisons solubles, dans les eaux minérales, à l'état de traces ou de quantités parfois non négligeables. Ainsi les eaux du Mont Dore étaient qualifiées d'arsenicales.

Le clarke s'élève à 5 g/t[10] : l’arsenic n'est pas un élément très rare. Il est présent dans de nombreux autres minéraux, particulièrement les arséniures, les arséniates, quelques sulfosels (alloclasite, cobaltite, énargite, lautite, luzonite, pearcéite, proustite, etc.).
Gisements exploitables
Le minerai principal est toutefois le composé minéral mispickel, une arsénopyrite de fer où le soufre et l'arsenic peuvent se substituer. L'extraction du mispickel (FeAs et FeS2) semble ancienne : un chauffage en cornue cylindrique, avec ajout de fonte, s'imposait. L'arsenic corps simple volatil s'élevait dans le cylindre supérieur.
- FeAs solide arséniure de fer, associé dans le mispickel + FeS2 pyrite associée au mispickel chauffé → As vapeur volatile captée puis dépôt sur surface froide + FeS masse solide plus dense
Une distillation sur charbon actif permettait de purifier la matière arsenic.
Accessoirement, il est possible de considérer le réalgar AsS (« arsenic rouge ») et l'orpiment As2S3 (« arsenic jaune ») comme d'autres minerais.
Flux naturels
Il s'agit d'un oligoélément à très faible dose, mais d'un poison toxique puissant à doses plus élevées.
Le cycle de l’arsenic est mal connu, mais on estime que les bactéries terrestres produisent environ 26 000 t/an d’arsenic méthylé volatil aboutissant à l’océan. S’y ajoutent 17 000 t/an émises par les volcans et 2 000 t/an environ issues de l’érosion éolienne des sols. Les sédiments marins en piègent une partie, qui durant un certain temps restera toutefois biodisponible. Ces flux sont à comparer à la production par les activités humaines, qui étaient estimées dans les années 1990 à environ 30 000 t/an dans le monde.
Une faible quantité d’arsenic est présente dans tous les organismes marins. Pour des raisons encore mal comprises, les invertébrés et en particulier les mollusques bivalves (moules, huîtres, coquilles Saint-Jacques) de pleine mer sont souvent plus contaminés (10 à 30 µg/L en général) que ceux qui vivent dans les estuaires. Des pics de pollution marine peuvent être constatés, par exemple autour des sites de munitions immergées en mer Baltique.
Des taux élevés (par exemple 2 739 µg/L chez le polychète Tharyx marioni sont constatés dans les zones très polluées. Chez les poissons, des concentrations de 5 à 100 µg/L sont courantes, plus faibles chez les espèces consommant du plancton (bar, maquereau, hareng) (5 à 20 µg/L) et plus élevées chez les poissons du sommet du réseau trophique (130 et 230 µg/L respectivement pour le congre et la roussette).
Les teneurs des poissons plats (plie, sole, flet) variaient de 10 à 60 µg/L en France dans les années 1990[11].
Corps simple
Propriétés physiques
L'arsenic solide se présente sous trois formes allotropiques dites arsenic jaune, arsenic noir et arsenic gris.
L'arsenic jaune, de formule As4, est non métallique et de structure tétraédrique. On l'obtient par condensation rapide de vapeur d'arsenic. C'est l'analogue de P4 et surtout de Sb4.
L'arsenic noir, parfois qualifiée de semi-métallique, est plus stable. C'est l'analogue de l'antimoine noir et du phosphore noir. Le reflet est nettement métallique, il est modérément conducteur de la chaleur et de l'électricité. C'est un solide cassant de densité avoisinant 5,7 (5,725 pur), de couleur gris de fer à gris métal, doué d'éclat métallique.
L'arsenic chauffé vers 300 °C commence à se sublimer sans fondre. La sublimation est rapide et totale à 613 °C. Les vapeurs se condensent sur les parois ou les surfaces plus froides, en formant petit à petit des rhomboèdres. Le chimiste allemand Mitscherlich a mesuré une densité de vapeur d'As avoisinant 10,37 sous la pression de vapeur non contrainte d'un volume molaire, autant à 564 °C qu'à 860 °C.
Le point de fusion peut être atteint sous de plus grandes pressions, de l'ordre de 36 atmosphères, à 817 °C. On peut faire fondre l'arsenic en le chauffant dans un tube scellé à la lampe à souder, dont la partie inférieure est en forme de canon de pistolet. Cette forme empêche la déformation du verre sous la pression de vapeur croissante.
Une synthèse des résultats expérimentaux concernant le diagramme de phase de l'arsenic est publiée en 1989[12] :
- le point triple se situe à TT = 817 °C et PT = 36,3 ± 0,5 bar (35,8 ± 0,5 atm) ;
- l'arsenic solide à l'équilibre est As-α, rhomboédrique (symbole de Pearson hR2 ; groupe d'espace R3m). À 14 °C, sa masse volumique est de 5,73 g/cm3, et donc son volume molaire de 13,08 cm3/mol. Le paramètre cristallin a ne dépend pas significativement de la température jusqu'à TT, alors que c suit la loi c (nm) = 1,0534 × [1 + 4,747 × 10−5 t (°C)] : le volume molaire est donné par V (cm3/mol) = 13,071 [1 + 4,747 × 10−5 t (°C)]. À haute pression (vers 120–150 bar à température ambiante) le solide devient tétragonal (a = 0,869 1 nm et c = 0,636 3 nm, c/a = 0,732 ± 0,001). À 140 kbar, l'arsenic tétragonal devient supraconducteur en dessous de 0,5 K ;
- équilibre solide-gaz : au-dessous de PT le solide chauffé se transforme directement en gaz (sublimation). À pression ambiante le point de sublimation est à 614 ± 1 °C. L'arsenic gazeux est essentiellement constitué de tétramères As4, mais comporte aussi As, As2 et As3 (au total moins de 0,19 % à l'équilibre avec le solide à 614 °C, moins encore à plus basse température) ;
- équilibre solide-liquide : au-dessus de PT le solide chauffé se transforme d'abord en liquide. La pente de la courbe d'équilibre vaut dT/dP = 0,003 6 K/bar. L'enthalpie de fusion vaut 23,848 kJ/mol. De TT à 890 °C, la masse volumique du liquide à l'équilibre avec le solide varie avec la température selon la loi ρ (g/cm3) = 5,80 − 0,000535 T (K).
Propriétés chimiques
L'arsenic insoluble dans l'eau se ternit à l'air, il se couvre alors de poussières noires. Il est possible de nettoyer poussières et surface altéré à l'eau de chlore.
L'arsenic chauffé dans l'oxygène raréfié, c'est-à-dire le gaz dioxygène à faible pression partielle, sous cloche, est phosphorescent. Si la température s'accroît ou la pression partielle d'oxygène s'élève, l'arsenic brûle avec une flamme verdâtre.
Projeté sur des charbons ardents, l'arsenic se volatilise avec une odeur d'ail forte. La vapeur est très sensible à l'oxydation chimique, et très vite, de l'anhydride arsénieux As2O3, voire de l'acide arsénieux en milieu humide, apparaît sous forme de dépôts.
L'arsenic traité par l'acide nitrique donne de l'acide arsénique présent uniquement en milieu aqueux. Cette réaction oxydante lente est parfois décrite comme une dissolution.
- 3 As solide cristal + 5 HNO3 aq acide fort concentré + 2 H2O → 3 H3AsO4 aq acide arsénique + 5 NO gaz
Cette réaction avec l'acide nitrique est analogue à celle du corps simple P.
Il est plus aisé d'oxyder le corps simple As en portant l'élément à son état d'oxydation le plus élevé, en solution basique qu'en solution acide.
Projeté en poudre dans un flacon rempli de gaz dichlore, l'arsenic brûle avec une flamme blanche et laisse un dépôt de AsCl3. La réaction a également facilement lieu avec les autres halogènes. Ainsi peuvent être produits directement le bromure AsBr3, les iodures AsI3 et AsI5, voire les fluorures AsF3 et AsF5.
Composés chimiques
Composés inorganiques
L'oxyde d'arsenic ou anhydride arsénieux As2O3 est amphotère, à l'instar de l'oxyde d'antimoine, ce qui atteste, pour les chimistes, un caractère semi-métal de ce métalloïde. Il existe sous trois formes allotropiques. Sa morphologie peut être amorphe ou vitreuse, ou encore cristalline, soit cubique soit monoclinique.
Le trioxyde d'arsenic, anhydride arsénieux, ou encore « arsenic blanc », improprement appelé arsenic, de formule As2O3, est un poison violent. Il est néanmoins indiqué et investigué comme anticancéreux dans certaines leucémies[13] - [14] ; ses effets indésirables incluent troubles hydroélectrolytiques, arythmie cardiaque, voire l’arrêt cardiaque pouvant entraîner la mort.
Il est utilisé pour la fabrication de verre ou de cristal, quand il n’a pas été remplacé par le trioxyde d’antimoine, également toxique, mais non soumis à la directive Seveso.
L'acide arsénique ou arséniate As2O5 existe également.
La plupart des composés arsenicaux dérivent notamment de l'acide arsénique (H3AsO4) et des trois ions (acidité faible) : ion dihydrogénoarséniate (H2AsO4−), ion hydrogénoarséniate (HAsO42−), ion arséniate (AsO43−).
Les composés arsenicaux (organiques ou non organiques) sont des molécules surtout utilisées (ou autrefois utilisées car beaucoup sont maintenant interdites) comme biocides (fongicide, herbicide, insecticide, poisons raticides, etc.) ou qui ont été utilisées comme gaz de combat dans les armes chimiques développées durant la Première Guerre mondiale.
De manière assez similaire aux oxydes, il existe trois combinaisons communes avec le soufre : As4S4, As2S3 et As2S5
Les caractéristiques du gaz arsine AsH3 peuvent se rapprocher de la phosphine. Le trihydrure d'arsenic, arsénure d'hydrogène ou arsine, de formule AsH3, est une substance se vaporisant en un gaz incolore, d’odeur alliacée nauséabonde, très toxique qui a été utilisé comme gaz de combat, notamment lors de la Première Guerre mondiale. L'arsine disparaît en solution alcaline. Comme l'arsenic précipite, l'arsine peut être considéré comme un bon réducteur en milieu basique.
Le test de Marsh, qui consiste à verser de l'arsenic dans un mélange d'acide fort et de poudre ou fins copeaux de zinc, la réaction générant un flot de gaz arsine ou hydrogène arsénié, se déposant en miroir d'arsenic sur une surface préchauffée de verre ou de porcelaine s'applique à la réduction d'un composé d'arsenic (ou d'antimoine). Mais le miroir d'arsenic est facilement dissous par une solution d'hypochlorite de sodium, contrairement à celui d'antimoine.
Les autres sels et composés inorganiques sont le plus souvent :
- des sels trivalents (As (III), As+3 ; formes variées d’arsénites, dérivées du trioxyde d'arsenic en solution aqueuse),
- des sels pentavalents ((As (V), As+5) ; ou arséniates, dérivés du pentoxyde d'arsenic en solution aqueuse), qui sont produits à base de trioxyde d'arsenic, d'acétoarsénite de cuivre (« Vert de paris », Paris Green), d'arséniate de plomb, ou d'arséniate de calcium.
Composés organiques
Ce sont notamment l’arsénobétaïne et l’arsénocholine, notamment présents dans diverses plantes et organismes marins. Ce sont aussi les métabolites de l’arsenic :
- dérivés mono et diméthylés de l'arsenic ;
- l'acide monométhylarsineux (AMMA (III)) ;
- l’acide diméthylarsineux (ADMA (III)) ;
- l’acide diméthylarsonique (ADMA (V)) ou acide diméthylarsinique, ou acide cacodylique, encore dit « agent bleu », dont la forme de base est le cacodylate ;
- l’acide monométhylarsonique (AMMA V ou AMMA (V)) ;
- le sel monosodique de l’acide méthane arsénique ;
- le sel disodique de l’acide méthane arsénique, très utilisé dans les désherbants les plus courants des golfs nord-américains ou pour défaner le coton[15].
Aux doses habituellement présentes dans la Nature ils sont moins toxiques pour les animaux à sang chaud, mais ils contiennent néanmoins de l'arsenic non dégradable.
En médecine, ils ont remplacé les sels d'arsenic, trop dangereux pour l'être humain et les animaux domestiques[16] et souvent maintenant interdits (sauf exceptions dans certains pays comme les États-Unis, durant un certain temps, comme les produits arséniés de traitement du bois, l’arséniate de plomb comme régulateur de croissance pour le raisin, ou l'arséniate de calcium pour les gazons de golfs qui ont été autorisés par l'EPA aux États-Unis après que les autres arsénicaux non organiques eurent été interdits). On les trouve notamment dans certains herbicides très utilisés sur les golfs nord-américains ou pour le défanage du coton avant récolte[15].
L'arsénite de soude n'a été en France interdite sur la vigne comme pesticide (insecticide utilisé contre la pyrale de la vigne, les carpocapses les vers des pommes et des poires, des altises, la pyrale, la cochylis et l’eudémis puis comme fongicide) que le par le ministère de l'Agriculture sous l'égide de Jean Glavany et en 2004 par la Commission européenne[17]. Peu après, des viticulteurs soutenus par la FNSEA réclamaient sa réautorisation pour lutter contre l'esca (aussi dit maladie du bois de vigne, d'abord attribuée à deux basidiomycètes, mais qui serait due à un complexe d'au moins cinq organismes)[18]. Des composés arsénicaux inorganiques interdits en Europe ou aux États-Unis peuvent encore être utilisés comme pesticides dans certains pays, et ils ont de nombreux autres usages dans le monde entier[15].
- Durant la Seconde Guerre mondiale (octobre 1943), la réglementation s'intéresse à l’arsenic dans les raticides et taupicides, dans lesquels l'arsenic sera peu à peu remplacé par des anticoagulants, de la strychnine, des anti-vitamines K ou des sels de thallium en mélange avec divers ingrédients.
- En 1973, un décret interdit en France tous les herbicides arsenicaux, mais ils restent très utilisés aux États-Unis.
Histoire
Son nom vient du syriaque ܠܐ ܙܐܦܢܝܐ (al) zarniqa, issu du persan زرنيخ zarnikh signifiant « jaune » puis « orpiment » (sulfure naturel d'arsenic servant à peindre la peau des êtres humains sur les fresques de la Grèce antique). Le terme est adopté dans la langue grecque sous la forme ἀρσενικόν / arsenikón, qui correspond en étymologie populaire à la forme neutre du grec ἀρσενικός / arsenikós, « qui dompte le mâle » en raison de sa forte toxicité. Le terme grec est lui-même adopté en latin sous la forme arsenicum, qui donne en français arsenic[19]. Le prénom Arsène est tiré de la même racine grecque arsen (« mâle »).
L'adjectif arsenical au singulier, arsenicale au féminin, arsenicaux au pluriel qualifie un corps ou une matière qui contient de l'arsenic.
À l'âge du Bronze ancien le bronze est souvent composé d'un alliage à base de cuivre et d'arsenic, ce pourquoi les archéologues nomment parfois cette période l'âge du Bronze-Arsenic : employé comme durcissant et pour augmenter la brillance du métal, cet arsenic, selon les cas est une impureté naturelle du minerai de cuivre ou il a été ajouté intentionnellement comme adjuvant. Au Bronze final se substitue à ce bronze arsénié un alliage cuivre-étain permettant de fabriquer des métaux plus résistants et ductiles (âge du Bronze-Étain)[20].
Durant l'Antiquité, l'arsenic est toujours utilisé pour la métallurgie (durcissant de nombreux métaux) mais aussi dans les arts (pigments, peinture) et la médecine sous deux formes inorganiques à l'état naturel, du tri sulfure d'arsenic (l'orpiment As2S3) et du quadrisulfure d'arsenic (réalgar As4S4)[21]. Hippocrate les utilise au Ve siècle av. J.-C. pour soigner les ulcères cutanés. Depuis lors, la pharmacopée grecque et chinoise s'en sert pour traiter ou freiner la syphilis, le cancer, la tuberculose ou le paludisme[22].
Au VIIIe siècle, l'alchimiste arabe Jabir Ibn Hayyan est probablement le premier à préparer le trioxide d'arsenic en l'isolant de son composé minéral : cette poudre blanche sans goût et sans odeur la rendra indécelable jusqu'au XXe siècle car elle donne les mêmes symptômes que des intoxications alimentaires, ce qui lui confère le titre de « poison des rois et roi des poisons »[23].
On attribue à Albert le Grand d'être le premier à avoir isolé l'élément arsenic en 1250[24].
Au XVIIe siècle l'arsenic va être utilisé comme poison sous le nom de « poudre de succession » par un réseau composé essentiellement de femmes de la noblesse dans le but d'accélérer certains héritages par des meurtres[22].
Le liquide fumant de Cadet (composé As2(CH3)4O préparé en 1760 par le chimiste Louis Claude Cadet de Gassicourt) est le premier composé organométallique à avoir été synthétisé par l'être humain[25].
Le vert de Scheele à base d'arséniate de cuivre, inventé par Carl Wilhelm Scheele en 1775, remplace comme pigment vert le carbonate de cuivre. Pigment de peinture, il colore les papiers peints, les jouets d'enfants puis est remplacé par le vert de Schweinfurt tout aussi toxique[26].
La liqueur de Fowler à base d'arsénite de potassium découverte en 1786 par Thomas Fowler est utilisée comme remède et tonique pendant plus de 150 ans.
Les composés à base d'arsenic sont utilisés dans les teintures, l'oxyde d'arsenic jouant le rôle de mordant, pigments de réalgar ou d'orpiment, leur toxicité étant à l'origine — qui se révèle fausse en fait — du bannissement de la couleur verte au théâtre[27].
À partir de 1740, on le retrouve dans les traitements des semences à l'arsenic (tels le vert de Paris utilisé comme insecticide ou raticide) mais sa toxicité entraîne son interdiction dans cette industrie en 1808[28].
En 1908, Paul Ehrlich met au point un composé arsenical, le Salvarsan, considéré comme le premier agent anti-infectieux et chimiothérapeutique.
Il a été utilisé pour augmenter la toxicité de certaines armes chimiques (dont l'Ypérite), sous forme d’arsine notamment, dès la Première Guerre mondiale. La destruction d'armes chimiques après les opérations de désobusage a été source de pollutions durables[29]. Sous forme d’arsine il a été présent dans certaines munitions chimiques de la Première Guerre mondiale et des années qui ont suivi (fabriquées, non utilisées puis démantelées ou jetées en mer). Les arsines sont employées comme arme chimique durant la Première Guerre mondiale, notamment celles chargées dans des obus à « croix bleue ». En France, durant la Première Guerre mondiale, en 1916, l'arsenic est utilisé dans les armes chimiques. Pourtant, pour éviter dans le civil que ces produits soient utilisés comme poison contre des humains, un décret précise que « Article 1er : les composés arsenicaux insolubles destinés à la destruction des parasites de l’agriculture ne peuvent être vendus ni employés en nature. Ils doivent être mélangés avec une substance odorante et colorée en vert, suivant la formule indiquée à l’article ter de l’article ci-après » (décret du 15 décembre 1916).
« Les composés arsenicaux destinés à la destruction des parasites nuisibles à l’agriculture ne peuvent être délivrés ou employés pour cet usage qu’à l’état de mélange avec des dénaturants d’après la formule suivante • produits arsenicaux insolubles 1 000 g • pyridine ou phénol brut ou nitrobenzine : 20 g • vert sulfoconjugué : 2 g », mélange devant être tout à fait homogène. De plus, pour limiter les risques de détournement, le gouvernement impose que tout commerce de préparations arsenicales doit « avoir un registre coté et paraphé par le Maire ou le Commissaire de police. Toute préparation arsenicale doit être inscrite sur ledit registre »[30].
Les peintures anciennes exposées à la lumière dans les musées et qui utilisent des pigments contenant de l'arsenic, tels l'orpiment, voient ce composé photo-oxydé en anhydride sulfureux qui rend la peinture cassante et en trioxyde d'arsenic (employé jadis comme mort aux rats, ce composé n'est pas libéré en quantité suffisante pour être dangereux pour l'être humain) qui donne à la toile une teinte blanchâtre, d'où la nécessité de poser des filtres sur les fenêtres des salles des musées actuels[31].
Sous forme d’arséniate de plomb notamment, il a été utilisé comme pesticide, qui a été une source fréquente d’empoisonnement des utilisateurs ou de consommateurs de produits traités. Il continue à polluer l’environnement longtemps après son utilisation, le plomb et l’arsenic n’étant pas biodégradables ni dégradables à échelle humaine de temps.
Utilisations
- C'est l'agent de l’arsenicage qui consiste à immerger des cuirs et peaux dans une solution d'arséniate de soude pour éliminer les parasites (dermertes, anthrènes) : ces peaux arseniquées n'empêchent pas la prolifération de bactéries de putréfaction[32].
- Additionné au plomb, avec un peu d'antimoine, à raison de 5 à 8 %, il durcit légèrement la grenaille de plomb des cartouches de chasse (encore autorisées et utilisées en France, hormis dans les zones humides et vers les zones humides où elles sont interdites). Sans cela, les billes de plomb, s'écraserait les unes contre les autres au moment du tir et se déformeraient en perdant de leur énergie cinétique et de leurs qualités balistiques. Il sert aussi de durcisseur au plomb des cartouches munitions de guerre. Dans les deux cas, il freine aussi la formation d’oxyde de plomb.
- Additionné au mélange plomb-antimoine des électrodes, il améliore le fonctionnement des accumulateurs.
- Mélangé avec du cuivre et du chrome (CCA) c’est un produit de traitement du bois (qui lui donne une couleur verdâtre). Bien que controversé en raison de sa toxicité, de sa rémanence et du fait qu’il soit partiellement soluble dans l’eau et les pluies, ce traitement reste autorisé dans la plupart des pays.
- Il est depuis peu utilisé sur les « tambours » des imprimantes dites « lasers » et des photocopieuses, fax. Pur ou sous forme de séléniure, sa sensibilité à la lumière permet de décharger la tension électrostatique qui retient le toner.
- Les alliages composés d’arsenic et de gallium (GaAs) ou d’indium (InAs) donnent des matériaux semi-conducteurs (dits III-V par référence aux colonnes de la table des éléments), utilisés pour la fabrication de cellules photovoltaïques, de diodes électroluminescentes (DEL) et de transistors à très haute fréquence.
Plus chers et de mise en œuvre plus complexe que le silicium, leur marché reste marginal, mais leur rôle est essentiel en opto-électronique, où les performances du silicium sont moins bonnes. - Les insecticides anti-fourmis en contiennent souvent (dimethylarsinate de sodium), bien que l'usage de celui-ci soit interdit dans l'Union européenne.
- La roxarsone (commercialisée aux États-Unis sous le nom « 3-Nitro »[33]) est un antibiotique organo-arsénié. Il est utilisé depuis les années 1970 (récemment interdit en Europe, mais encore utilisé dans de nombreux pays, dont aux États-Unis comme additif alimentaire dans l'alimentation animale) pour traiter les maladies causées par les coccidies et pour éviter les « troubles de croissance » des porcs et volailles (hormis dans le Maryland où il est interdit depuis 2013[34]). Selon une étude récente, cet arsenic inorganique serait particulièrement concentré dans les poulets industriels traités aux antibiotiques. Le laboratoire Pfizer a annoncé, en 2011, la suspension de la vente[35], mais malgré les protestations d'associations environnementalistes ou de consommateurs, il est encore en 2013 autorisée par la FDA. Les inventeurs de ce produit affirmaient qu'il serait excrété sous la même forme[36]. On a ensuite montré que les oiseaux peuvent métaboliser l'arsenic organique en une forme inorganique beaucoup plus toxique[36]. De plus, alors que les excréments frais de volaille ne contiennent presque que la forme organique de l'arsenic ; une fois dans le sol, les bactéries le transforment en arsenic inorganique très toxique, retrouvé dans les eaux de ruissellement et les sédiments[37]. Les excréments de volailles traités à la roxarsone épandus comme engrais la péninsule de Delmarva ont libéré assez d'arsenic pour polluer le sol au-delà des normes fédérales de remédiation (sans toutefois à ce jour affecter la nappe)[37]. Une étude de la FDA a montré que les foies de volailles ayant consommé de médicament contenait plus d'arsenic que ceux d'animaux n'en ayant pas consommé[36]. Pourtant, selon Christopher Loder (porte parole de Pfizer), ce laboratoire n'aurait « pas connaissance de preuves démontrant qu'utiliser des roxarsone est source de risque pour l'environnement ».
Méthodes d'analyse
Plusieurs méthodes sont employées, dont :
- quantification de l’arsenic total d’un échantillon ;
- étude de la spéciation de l’échantillon, c’est-à-dire des différentes formes chimiques ou organiques d’arsenic présentes et quantification ;
- analyse de l’arsenic parmi d’autres métaux, via par exemple l’analyse par fluorescence X.
Analyse des échantillons d'eau
Une bonne méthode d’analyse doit avant tout minimiser les interférences de manière à être suffisamment sensible pour obtenir de bonnes limites de détection et de quantification. Ces limites doivent être inférieures aux normes nationales en vigueur. De plus la méthode doit être validée en termes de domaine de linéarité, de réplicabilité, de répétabilité et de justesse. Il est également important de connaître le taux de récupération pour ajuster les résultats.
Prélèvement et conservation de l'échantillon
L’analyse de l’arsenic étant généralement une analyse de trace il est indispensable de prélever les échantillons dans des contenants préalablement lavés avec de l’acide nitrique ou de l’acide chlorhydrique et rincés à l’eau déminéralisée. Pour garantir la conservation de l’échantillon, celui-ci devra être acidifié et ne pas être en contact avec l’air.
Techniques courantes en laboratoire
Il existe une large gamme de méthodes pour analyser les échantillons liquides contenant l’arsenic. Le choix de la méthode se fait en fonction des limites de détections désirées et de la concentration attendue. Parmi elles :
- la colorimétrie : peu précise ;
- la spectrométrie d'absorption atomique de flamme (FAAS) : très interférée et manque de sensibilité ;
- la spectrométrie d’absorption atomique avec atomisation électrothermique (GF- AAS). La mesure se base sur l’injection directe de l’échantillon dans un tube graphite, chauffé électriquement avec atomisation électrothermique ;
- la génération d’hydrure suivie d’une détection par spectroscopie d'absorption atomique (HG-AAS) ou par fluorescence atomique (HG-AFS). Dans un premier temps l’As(V) est réduit en As(III) avec de l’iodure de sodium. L’arsenic est ensuite transformé en un arsine volatil sous l’action du borohydride de sodium (NaBH4) en milieu acide. L’arsine formé est ensuite oxydé en arsenic élémentaire dans une cellule chauffée et dosée par spectrophotométrie d’absorption atomique. ;
- la spectrométrie d'émission atomique dans un plasma d’argon (ICP-AES) (voir Torche à plasma) ;
- la spectrométrie de masse par torche à plasma (ICP-MS). L’échantillon est introduit dans un plasma d’argon, cela permet son ionisation, le spectromètre de masse sépare ensuite les ions en fonction du rapport m/z (masse/charge). Cette méthode comporte de nombreux avantages : étant donné qu’il n’y a qu’un seul isotope stable pour m/z = 75, la détermination de l’arsenic est simple. De plus les limites de détection sont très faibles. Par contre, il y a un problème d’interférences isobariques dans les échantillons à concentration élevée en chlorure (présence de ArCl). De plus le coût d’investissement est élevé et l’utilisateur doit être compétent.
Méthodes d'analyse sur site
L’EPA a validé quelques méthodes d’analyse sur site[38]. Les trois premières sont basées sur la génération d’arsine. La mesure se fait par comparaison du changement de couleur de la solution avec les échelles de couleur fournies avec le kit.
- Peters Engineering As 75 PeCo test kit.
- Envitop Ltd. As-Top Water arsenic test kit.
- Industrial Test Systems, Inc., QuickT test kit arsenic analysis systems.
Le quatrième kit utilise une méthode voltamétrique.
- Nano-Band Explorer Portable Water Analyzer.
Des analyseurs portables (il existe aussi des appareils de fluorescence X portables, plus coûteux)
Extraction de l'échantillon
Les échantillons sont généralement préalablement séchés et tamisés. On procède ensuite à leur solubilisation soit par attaque acide sur plaque ou à reflux, soit par attaque acide en micro-ondes fermé pour ne rien perdre. Il n’existe cependant pas de méthode normalisée pour l’arsenic dans les sols et sédiments. L’échantillon est alors sous forme liquide et acidifiée.
On peut de même procéder à une analyse directe de l’échantillon sans minéralisation préalable.
Méthodes courantes en laboratoire
- Mêmes méthodes que pour l’analyse de l’eau après la mise en solution des échantillons.
- L’activation neutronique instrumentale (INAA) : utilisée comme technique de référence mais nécessitant l’accès à un réacteur nucléaire.
- La fluorescence X et ses contraintes : préparation des échantillons, étalonnage et prise en compte des effets de matrice.
Spéciation
Biométhylation : on a montré[39] en microcosmes, mésocosmes[40] et en laboratoire que dans le sol, l'eau, les sédiments ou le tube digestif, les microbes peuvent méthyler ou déméthyler l'arsenic, et transformer des espèces inorganiques de l'arsenic en formes organiques, et inversement[39].
Enjeux : la connaissance de la spéciation de l’arsenic permet de mieux mesurer l’impact et le risque environnemental, car la nature de l'espèce chimique affecte la biodisponibilité, la toxicologie et la mobilité et bioturbation de l’arsenic[41].
Implications analytiques : une analyse physico-chimique complète d'un environnement implique l'identification et quantification de nombreuses espèces. Or celles-ci peuvent assez rapidement évoluer, la stabilité des échantillons est donc importante, du prélèvement à l’analyse. L’extraction de l’arsenic ne doit pas modifier ses formes chimiques. Les techniques d’analyse doivent être sensibles, sélectives et rapides pour éviter la conversion des espèces présentes.
Stabilité de l'échantillon
Elle peut être obtenue en ajoutant un agent chélatant comme l’EDTA (acide éthylènediaminetétraacétique), et en réfrigérant les échantillons (à température ambiante, seules les solutions très concentrées restent stables).
La stabilité dépend de la nature de l’analyse : l’arsenic organique (méthylé) est plus stable que l’arsenic inorganique.
Extraction
Pour extraire l’arsenic sans modifier ses formes chimiques, il faut un solvant n’interférant pas avec la détection.
Le lessivage des sols permet de se rendre compte de la mobilité de l’arsenic (qui dépend de la composition chimique du sol, du pH, des micro-organismes, etc.). Pour extraire rapidement l’arsenic, des solvants forts sont utilisés et ils interfèrent ensuite avec la séparation HPLC.
Techniques de séparation
La chromatographie en phase liquide à haute performance (CLHP) est la technique la plus souvent utilisée. Ainsi on utilise la chromatographie de paire d’ions pour séparer les espèces neutres des espèces ioniques (cations ou anions), la chromatographie à échange d'ions (échange d’anions pour séparer As(III), As(V), MMA, DMA, de cations pour séparer l’arsénobétaïne, l’oxyde triméthylarsine et Me4As+).
La chromatographie d'exclusion stérique peut également être utilisée en tant que technique préparative.
L’électrophorèse capillaire est peu utilisée à cause des interférences dues à la matrice de l’échantillon. Cette technique est utilisée pour l’analyse de standard ou d’échantillons dont la matrice est simple.
Techniques de détection
Les principales techniques utilisées sont :
- la spectroscopie d'absorption atomique et l’ICP-AES. Ces techniques sont efficaces pour des échantillons très concentrés mais pas assez sensibles pour l’analyse de trace. Il faut alors les combiner avec la génération d’hydrure. La génération d’hydrure est une réaction rapide qui augmente de dix à cent fois la sensibilité. Cela permet aussi de supprimer l’effet de matrice de l’échantillon ;
- ICP-MS. Cette technique est ultrasensible et permet d’analyser différents éléments en même temps. De plus le couplage avec l’HPLC est facile et il est possible de mesurer les ratios isotopiques ce qui rend la détermination des espèces plus précise ;
- la spectrométrie de masse avec une source électrospray. Cela permet une analyse directe ou couplée avec HPLC et ajoute le paramètre identification des espèces arséniées en plus de les quantifier. Cela est particulièrement intéressant pour les molécules organiques complexes.
Toxicologie


Facteurs de toxicité
La toxicité de l’arsenic[43] dépend de sa nature chimique : l'arsenic inorganique est beaucoup plus toxique que l'arsenic organique (son niveau de toxicité dépend aussi de son degré d’oxydation : As(0) > As(III) > As(V)[44]).
L'arsenic est dit inorganique quand il est sous sa forme pure ou qu'il est lié à l’oxygène, au chlore ou au soufre. Il est alors très dangereux, même à faible dose, surtout en cas d’exposition répétée. Il est dit organique quand il est chimiquement lié au carbone ou à l'hydrogène. Sous cette forme, il est toxique à forte dose mais nécessaire à faible dose pour le bon fonctionnement de l'organisme. C'est un « ultra oligo-élément » essentiel pour l’être humain, le poulet, la chèvre, le porc et quelques autres espèces. Les besoins pour l’être humain ont été évalués entre 10 et 20 µg/j. Ils sont largement couverts par l’alimentation[45] - [46].
Effets biochimiques majeurs
- L'arsenic découple la chaîne respiratoire en se substituant au phosphore (dans le phosphate, dans la réaction de formation de l’ATP).
- Les protéines coagulent quand la concentration en arsenic inorganique est forte : réaction arsenic/liens sulfures ou réaction arsenic/site actif.
- Il se complexe avec les groupes sulfhydryle des enzymes.
- C'est un perturbateur endocrinien.
- Il perturbe aussi la différentiation cellulaire[47], ce qui contribue à ses propriétés cancérogènes[48].
Imprégnation de la population
Elle varie beaucoup selon le contexte biogéographique et industriel, le métier, le mode de vie, l'alimentation (cf. consommation de fruits de mer, poissons marins carnivores ou gibier), et le contexte géologique et environnemental.
Du point de vue de la santé publique, l'embryon, le fœtus et la femme enceinte sont a priori plus vulnérables en termes de risques.
En France, le « Volet périnatal » du programme national de biosurveillance a évalué l’imprégnation des femmes enceintes notamment par l’arsenic (et d'autres métaux et quelques polluants organiques) à l'occasion du suivi d'une cohorte de 4 145 femmes enceintes (« Cohorte Elfe » comprenant des femmes ayant accouché en France en 2011 hors Corse et TOM)[49]. Le dosage urinaire[50] de 990 femmes enceintes arrivant à la maternité a confirmé l'omniprésence de l'arsenic dans l'environnement (et nos organismes). Les niveaux mesurés (moyenne géométrique à 11,0 μg/L, soit 15,1 μg/g de créatinine) sont proches des moyennes trouvées chez les femmes (enceintes ou non) en France et dans plusieurs pays ou régions d'Europe et d'Australie. Ils sont en revanche plus élevés que les niveaux mesurés aux États-Unis et au Canada au sein de la population générale adulte. Ce travail confirme une sur-imprégnation des Français par l’arsenic total (comparativement à l'Amérique du Nord), déjà montré en 2007 par un étude ENNS (Étude nationale nutrition santé mise en œuvre par Santé publique France), qui semble au moins en grande partie lié à une consommation plus élevée de produits de la mer qui sont des sources reconnues d'exposition à l'arsenic[49].
Toxicocinétique et métabolisation
Son degré d'absorption et de rétention (cinétique) par l'organisme, ainsi que sa modification (métabolisation) dans l'organisme vont fortement dépendre[51] :
- de sa forme chimique, autrement dit de sa spéciation (espèce chimique) ; organique ou minérale) ;
- de sa valence (l'arsenic est souvent trivalent ou pentavalent dans l'environnement industriel) ;
- de sa forme physique (vapeur, nanoparticule, aérosol, particule, poussière, etc.) ou de sa granulométrie voire de la forme du fragment s'il s'agit d'un morceau d'arsenic ou de ses composés ;
- de la voie d'incorporation ; sa métabolisation et son élimination ou transfert vers certains organes se feront différemment selon la voie d'incorporation (orale, pulmonaire, percutanée, etc.).
À titre d'exemple, en moyenne et dans un milieu de travail exposé à ce risque, 80 % environ de l'arsenic ingéré passe dans le sang contre 40 à 60 % de l'arsenic inhalé sous forme de poussières et vapeurs[51]. Une faible contamination percutanée existe aussi[51].
Distribution : elle se fait rapidement, dès que l'arsenic a pénétré l'organisme. Ce dernier se fixe aux protéines et tend à s'accumuler dans le foie, la peau, les phanères et les poumons (où il peut être trouvé longtemps après la contamination alors que sa demi-vie sanguine, triphasique, est de 2-3 heures, 30 heures et 200 heures[51].)
Métabolisation : elle varie selon l'espèce chimique. Par exemple, l'arsenic pentavalent est métabolisé en arsenic trivalent, qui sera méthylé (méthylation oxydative avec formation d'acide monométhylarsonique (MMA). Ce MMA trivalent et hautement toxique sera ensuite transformé en acide diméthylarsinique (DMA) pentavalent et peu toxique.
En termes de médecine du travail, les métabolites recherchées dans les analyses d'urine sont généralement l'Arsénite de cuivre ; l'Arsénite de sodium ; le Pentoxyde de diarsenic ; le Trichlorure d'arsenic et le Trioxyde de diarsenic[51].
Demi-vie des métabolites : elle varie de deux à six jours (selon l'espèce chimique de départ).
Excrétion : 70 % des composés arsénieux inorganiques absorbés sont éliminés dans les urines. La moitié de l'arsenic est ainsi éliminé en 48 heures et 90 % en six jours[51]. Il l'est sous forme de dérivés monométhylés (et notamment sous forme d'acide monométhylarsonique ; 25 % du total environ), de dérivés diméthylés (acide diméthylarsinique pour 50 % environ) et sous forme inchangée pour le reste[51].
La bile transfère aussi de l'arsenic vers l'intestin.
Comme dans le cas du plomb et du mercure, le réseau sanguin en exporte aussi vers les phanères (poils, cheveux où il persiste jusqu'à leur chute et au-delà)[51].
Comme pour le plomb ou d'autres toxiques, il existe des différences interindividuelles de métabolisation[51], liées à la santé, l'âge et le sexe de l'individu, sans doute sur des bases au moins en partie génétiques.
Intoxication aiguë
Elle se traduit par des symptômes immédiats, dont des vomissements, des douleurs œsophagiennes et abdominales et des diarrhées sanguinolentes, entraînant le collapsus et la mort.
Intoxication par exposition chronique
L’arsénicisme[32] est l'exposition à de petites doses d'arsenic (via une eau de boisson polluée par exemple[52]).
Ses symptômes sont la mélanodermie, l'hyperkératose des mains et des pieds, l'alopécie et une polynévrite douloureuse, la striure des ongles.
Il est un facteur de risque de :
- cancer du poumon, inscrit à ce titre dans les tableaux de maladies professionnelles ;
- cancer de la peau[53] où l'arsenic tend à s'accumuler en cas d'exposition chronique, et où il est principalement source de la maladie de Bowen[53] (carcinome in situ), Carcinome basocellulaire (CBC)[53] et Carcinome épidermoïde[53] ;
- cancer de la vessie ;
- cancer du rein ;
- athérosclérose (notamment de la carotide)[54], ou responsable d'autres maladies cardio-vasculaires[55]
- probablement de maladies respiratoires[56].
- dépression immunitaire[57].
Les premières manifestations visibles sont généralement cutanées, avec une augmentation de la pigmentation. Le cancer survient plus tardivement et peut mettre plus de 10 ans à apparaître.
Selon Manote, l’absorption d’arsenic par la peau ne présenterait paradoxalement apparemment pas de risque pour la santé[58].
L’arsenic est souvent employé comme poison, d’où le titre Arsenic et vieilles dentelles. Certains chercheurs supposent que Napoléon Ier aurait été empoisonné à l’arsenic, en raison de la forte concentration en arsenic dans ses cheveux (l'arsenic tend à s'accumuler dans les phanères du corps), cependant, l'arsenic était aussi utilisé à cette époque comme agent de conservation, d'où les doutes à propos de cet empoisonnement[58] ; diverses affaires judiciaires ont été liées à l'empoisonnement à l'arsenic, notamment l'affaire Marie Lafarge et l'affaire Marie Besnard.
Perturbation endocrinienne
L'arsenic est aussi un puissant perturbateur endocrinien[59] - [60] - [61]. Cet effet sur la santé est documenté chez l'être humain et sur le modèle animal en laboratoire dès 10 à 50 ppb, et à des doses encore bien plus faibles sur culture cellulaire, ce qui en fait un contaminant préoccupant de l'eau potable et de l'environnement dans certaines régions du monde[59].
Il perturbe les récepteurs stéroïdes des androgènes, de la progestérone, de minéralocorticoïdes et glucocorticoïdes, ainsi que la régulation des gènes à des taux aussi bas que 0,01 µM (environ 0,7 ppb)[59]. De très faibles doses ont renforcé la transcription des gènes dépendant d'un médiateur hormonal, tandis que des doses légèrement plus élevées mais non cytotoxiques l'ont inhibée, in vivo et dans les cultures cellulaires[59]. Ce pourrait être l'une des explications de ses propriétés cancérigènes[59]. Chez le rat les analyses histologiques et moléculaires montrent que l'arsenic inhibe le développement de la prostate à la prépuberté, « en compromettant la maturation structurelle et fonctionnelle de la prostate chez les rats pubertaires aux deux doses évaluées dans cette étude » (au moins dès 0,01 mg de NaAsO2 par litre d'eau de boisson à la prépuberté et dès 10,0 mg/L à la puberté)[62].
Écotoxicologie
L'arsenic est un oligo-élément à très faible dose, et certains organismes (champignons ou levures telles que Saccharomyces[63] et bactéries notamment, mais aussi certaines plantes) disposent de mécanismes d'adaptation à sa toxicité à faible dose[64] - [65]. Mais dans l'environnement – où il est parfois un contaminant naturel des nappes phréatiques, au Bangladesh par exemple[66]– on le considère comme un polluant au-delà de seuils d'écotoxicité, qui varient selon sa forme chimique et la nature des sols (la teneur du sol en humus et en fer contrôlent fortement sa biodisponibilité)[67] - [68]. De plus, la toxicité de l'arsenic est liée à sa biodisponibilité et à sa bioaccumulation, deux facteurs qui dépendent du contexte pédologique, mais aussi de la spéciation de l'arsenic[69]. Comme pour d'autres toxiques tels que le mercure, cette spéciation est dans l'environnement réel très complexe (pour ses deux grandes formes ; organiques et inorganiques), en raison d'interconversions possibles et fréquentes entre les espèces chimiques, contrôlées par des processus à la fois biotiques et abiotiques[70].
Son caractère de perturbateur endocrinien est également démontré sur le modèle animal, mais avec des conséquences et niveaux d'effets mal évalués au niveau écosystémiques.
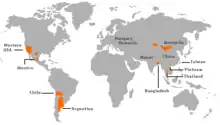
Carte des risques pour l'arsenic dans les eaux souterraines
Environ un tiers de la population mondiale consomme de l’eau potable provenant des nappes phréatiques. Un nombre approximatif de trois cents millions de personnes puisent leur eau dans des nappes phréatiques fortement polluées par de l’arsenic et du fluorure[71]. Ces éléments traces sont le plus souvent d’origine naturelle et proviennent des roches et des sédiments lessivés par l’eau. En 2008, l’Institut suisse de recherche de l’eau Eawag a présenté une nouvelle méthode permettant d'établir des cartes des risques pour les substances toxiques géogènes dans les eaux souterraines[72] - [73] - [74] - [75]. Cela permet de déterminer plus efficacement quelles sources devraient être contrôlées. En 2016, le groupe de recherche a mis ses connaissances en libre accès sur la plate-forme GAP (Groundwater Assessment Platform) (www.gapmaps.org). Celle-ci permet aux spécialistes du monde entier de charger leurs propres données de mesure, de les visualiser, et de créer des cartes des risques pour des régions de leur choix. La plate-forme sert également de forum d’échange de connaissances afin de contribuer au développement de méthodes visant à éliminer les substances toxiques de l’eau.
Origines et biodisponibilité
Elles sont liées au contexte et aux processus biogéochimiques et géochimiques[76] Son origine peut être naturelle, il est alors souvent associé à l'or (Au), à l'argent (Ag), au cuivre (Cu) et au sélénium (Se) en particulier[77] - [70]), mais la plupart du temps, sa présence à forte dose dans les sédiments ou le sol a une origine industrielle (mines, métallurgie, production de pesticides) ou agro-industrielle (pesticides arséniés massivement utilisés sur les rizières[70], les vergers[70], les cultures de coton[70], les golfs[78]).

De nombreux sols industriels sont pollués par de l'arsenic, de même que la nappe quand elle est présente. En France, par exemple, sur 4 142 sites pollués répertoriés par les données disponibles, « les sols de 488 d'entre eux (soit 12 %) sont pollués par l'arsenic, tandis que la pollution des nappes ne concerne que 257 sites pollués (soit 6 %) »[79].
L'arsenic est aussi parfois très présent dans les sols agricoles, voire dans certains jardins et vergers anciens, à la suite de son utilisation massive par plusieurs dizaines de marques de pesticides[68].
Ainsi, aux États-Unis, une étude faite dans l'État du New Jersey, basée sur les données publiquement disponibles au milieu des années 1990[68] a cherché à évaluer les quantités d'arséniates de plomb et de calcium épandus sur terrains de golf, gazons et champs de l'État du New Jersey. Les quantités épandues ont pu être estimés à 22 226 tonnes pour l'arséniate de plomb et 8 165 tonnes pour l'arséniate de calcium en 80 ans (de 1900 à 1980) ; soit un total cumulé équivalent à environ 6 804 tonnes d'arsenic pur pour cette période, ce qui a pu expliquer les teneurs élevées de plomb et d'arsenic des sols de cet État. Les auteurs de l'étude rappellent cependant que des évaluations de l'exposition humaine nécessitent que des données sur la consommation locale en ces pesticides soient rendues publiques[68].
Une des molécules actives très utilisées est le méthanearséniate monosodique (MSMA, épandu sur le coton pour le défanage chimique en pré-récolte, et sur les terrains de golf notamment). Cette molécule, de même que le méthanearséniate disodique, a remplacé l'arséniate de plomb[80] et l'arséniate de calcium[68] trop toxiques. C'est une molécule à la fois fongicide, désherbante et insecticide, très utilisée aux États-Unis (1 800 t/an) sur les champs de coton[81] et les golfs[78].
En Amérique du Nord, il a aussi été localement très utilisé en forêt pour traiter les résineux contre les invasions de scolytes, peut-être maladroitement, car il contribue à attirer les scolytes et à empoisonner leurs prédateurs.
Il s'agit parfois d'une séquelle de guerre. On a ainsi trouvé en Belgique en plein champ en lisière d'une zone forestière un site très pollué par le démantèlement après-guerre de munitions chimiques[82]. De même a-t-on trouvé jusqu'à plus de 100 mg/kg d'arsenic dans le sol et jusqu'à plus de 1 000 µg/L en pleine forêt de Verdun sur un ancien site de démantèlement de munitions de la Première Guerre mondiale. Une entreprise y avait brûlé durant la reconstruction le contenu chimique de plus de 200 000 obus chimiques allemands contenant notamment du chlorure de diphénylarsine et du cyanure de diphénylarsine)[83].
Tous ces usages sont sources de pollution durable du sol, de l'eau et des biotopes, dont la gravité pour la santé et les écosystèmes est encore discutée. Parce qu'il a été très utilisé comme pesticide et qu'il ne se dégrade pas, l'arsenic serait devenu le second polluant le plus préoccupant dans les sols aux États-Unis[84].
Chez les animaux
Hautement toxique autant pour l'être humain que pour l'animal, que ce soit à forte dose ou à faible dose, l'effet reste le même. L'animal qui l'ingère dans de la nourriture ne fera aucune différence et décédera dans les douze heures qui suivent.
Chez les végétaux
Des pollutions par l'arsenic sont fréquentes dans les zones minières[85] où il peut venir renforcer la toxicité d'autres contaminants (mercure autour des anciennes mines de ce métal, par exemple[86]). On le trouve aussi à l'intérieur et autour de certaines zones industrielles où il peut polluer les sols et contaminer les cultures alimentaires ainsi que les animaux consommés par l'être humain.
Au-delà d'un certain seuil de phytotoxicité[87] l'arsenic, par des processus encore incomplètement compris[88], modifie le métabolisme, inhibe la croissance, puis tue les plantes, monocotylédones comme dicotylédones[87].
Les plantes accumulent à la fois des formes organiques et inorganiques de l'arsenic, mais on ne sait pas encore clairement si elles peuvent ou non transformer des formes inorganiques en formes organiques. On sait qu'une complexation par la phytochélatine est utilisée par un grand nombre d'espèces végétales pour se détoxifier des formes inorganiques et potentiellement d'espèces organiques de l'arsenic[70]. Certains taxons ou variétés ont évolué en développant des résistances aux arséniates. S'il est connu depuis longtemps que l'arsénite et l'arséniate ont une action toxique par réaction avec des groupes-SH et en entrant en compétition avec le phosphate dans le métabolisme cellulaire, on a aussi montré (2002) que le stress oxydant causée par la réduction et l'oxydation cellulaire doit aussi être considéré comme une des explications de la toxicité de ces ions[70].
Au contraire de ce qui se passe pour les animaux, ce sont les formes non organiques de l'arsenic qui sont les plus toxiques pour les plantes, mais avec de fortes différences selon le type de sol[87]. Ces différences selon le sol sont expliquées par une biodisponibilité de l'arsenic liée à sa spéciation et à son degré d'adsorption sur l'argile ou les complexes argilo-humiques ou la matière organique du sol. La marge est parfois très étroite entre la teneur naturelle du fond géochimique et le niveau toxique pour la plupart des plantes[87].
Par exemple, l'arsenic inorganique (métalloïde) est cinq fois plus toxique dans les sables et les limons (moyenne géométrique (MG) des seuils de phytotoxicité signalés par la littérature 40 mg/kg) que dans les sols argileux (où la MG passe à 200 mg/kg)[87], ce qui explique qu'il n'y a pas de norme sol pour l'arsenic, car une norme pour l'arsenic dans les sols devrait être adaptée au type de sol[87], ce qui rendrait difficile son application.
- Certaines plantes (telles les fougères Pityrogramma calomelanos[89] ou Pteris vittata[90] ou Pteris cretica[91] (proposées pour la phytoremédiation) ont développé une certaine tolérance aux désherbants à base de dérivés organiques d'arsenic ; en réduisant l'arséniate en arsenite, et en translocalisant les toxines vers les parties aériennes (avec stockage vacuolaire)[92]. Ainsi, sur un sol contenant 97 ppm d'arsenic, les feuilles de P. Vittata (plante hyperaccumulatrice) peuvent (après 20 j de croissance) en bioaccumuler 7 000 ppm[93].
- Sur certains terrils et crassiers miniers riches en arsenic, certaines souches d'herbacées (« métallotolérantes » ou « métallophytes »), Agrostis tenuis notamment, survivent à des taux d'arsenic qui tueraient la plupart des plantes. Elles survivent à condition qu'il soit sous forme d'arséniate et non d'arsénite[94] - [95] et dans ce cas, l'arséniate est la forme la plus lixiviable de l'arsenic.
- En milieu aride, une Parkinsonia (Parkinsonia Florida) bioconcentre l'arsenic (deux fois mieux — dans ses racines — quand elle a poussé dans un sol sableux que dans un sol limoneux). Comme elle peut être cultivée, elle a été proposée pour la phytoremédiation de l'arsenic en région semi-arides[96].
- À Löcknitz, Allemagne, où persiste dans le sol une pollution qui est une séquelle de toxiques de combat arséniés manipulés durant la Seconde Guerre mondiale, des chercheurs ont montré que certaines plantes peuvent absorber des quantités significatives d'arsenic sans en mourir, pouvant ensuite contaminer le gibier ou le bétail. C'est le cas par exemple de la houlque laineuse (Holcus lanatus L.), une herbacée commune en Europe. Sur ce même site, des champignons prélevés sur des sols fortement contaminés ont montré des teneurs de 0,12 mg/kg[97].
- Des enjeux de santé environnementale forts existent car certaines plantes[98] dont en particulier le riz[99] peuvent bioaccumuler des quantités significatives d'arsenic[100] quand elles poussent dans de l'eau polluée, ce qui arrive facilement en aval des champs de coton. Les tomates[101], les haricots et de nombreuses autres plantes comestibles sont aussi concernées.
Certains organismes marins tels que les algues peuvent aussi bioconcentrer des quantités significatives d'arsenic sans en mourir[102]. Le cycle et la spéciation de l'arsenic a été étudié pour certains légumes, dont la carotte[103].
Dans le sol
Pratiquement toutes les formes de vie dans le sol[104] — sauf bactérienne extrêmophiles peut-être — semblent sensibles à l'arsenic, surtout à ses formes inorganiques. Certains champignons microscopiques le captent et pourraient, quand ils sont symbiotes de certaines plantes, faciliter ou au contraire (selon les cas) freiner les transferts d'arsenic du sol aux plantes[105], notamment dans les métallophytes[106], ou au contraire le bloquer hors du système racinaire dans le sol, semble-t-il via une protéine (la glomaline)[107], que certains auteurs préconisent de tester pour dépolluer des sols contaminés par des métaux[107]. C'est ainsi que la houlque laineuse ou quelques autres espèces résistent à des sols pollués par l'arsenic[108].
Une équipe de l'Institut d'astrobiologie de la NASA a publié un article controversé affirmant que la bactérie extrémophile (GFAJ-1) avait la capacité d'intégrer l'arsenic dans son organisme (y compris dans son ADN) à la place du phosphore qui est son analogue chimique (proche voisin dans le tableau de Mendeleiev)[109]. Cette découverte a cependant été invalide par une étude de 2012[110], qui a démontré :
- que cette souche ne pouvait survivre sans apport de phosphate, même en présence d'arsenic,
- que l'ADN de GFAJ-1 ne contenait que des traces d'arsenate, qui de plus n'étaient pas liées de façon covalente.
- L'écotoxicité d'un environnement enrichi en arsenic semble pouvoir être réduite par une forte teneur en oxyde de fer ou un apport de zéolite associés à un apport important en matière organique du sol[111].
- Un ajout de compost amendé avec de la zéolite et/ou de l'oxyde de fer (jusqu'à 20 % p/p) a été testé sur un sol fortement contaminé par l'arsenic (34 470 mg/kg de sol). La biodisponibilité de l'arsenic a ensuite été déterminée selon le taux d'arsenic absorbé par du ray-grass (Lolium perenne L.) récolté sur ce sol (cultivé sous serre). Les taux d'arsenic dans le ray-grass ont ainsi été réduits à 2 mg/kg (poids sec) que ce soit avec 15 % de compost comprenant 5 % d'oxyde de fer ou avec 15 % de compost enrichi de 5 % de zéolite[111]. Dans les deux cas, les plantes ont absorbé moins de 0,01 % de la teneur totale en arsenic du sol. Dans le sol enrichi en ces compost, la fraction soluble de l'arsenic a été réduite de 10 à 37 % et l'arsenic s'est redistribué principalement dans la matrice de fer et d'oxygène des échantillons traités[111].
- Il est parfois même présent dans les prairies[112], à la suite de retombées aériennes, d'apports d'effluents ou de ruissellement, ou enfin à la suite de l'utilisation antérieure de prairies pour le traitement des bois destinés à faire des piquets ou poteaux. Une étude néozélandaises a ainsi porté sur 50 échantillons de sols de prairies permanentes, collectés près de Levin, en 1989. Ces prairies à raygrass (Lolium perenne) et trèfle rampant (trifolium repens) avaient été diversement polluées par trois métaux (19 à 835 mg/kg de cuivre, 47 à 739 mg/kg de chrome, et 12 à 790 mg/kg d'arsenic dans les cinq premiers centimètres du sol), à la suite du traitement de bois de pin (pinus radiata).
Les analyses de sol ont montré que les teneurs en Cu, Cr, et As restaient corrélées entre elles, et qu'elles diminuaient avec la profondeur (mesures faites à 30 cm)[112].
Dans des parcelles moyennement à hautement contaminées, du coton enfoui ne se décomposait presque pas, preuve d'une inhibition de l'activité des décomposeurs. Un autre indice était la disparition totale des lombrics (Lumbricus rosea et Aporrectodea rubellus) dans les parcelles moyennement à très contaminées. Ailleurs, on n'a pas mis en évidence d'accumulation particulière de métaux lourds dans les tissus de vers de terre[112].
Le sol était plus compacté, et donc asphyxiant et moins drainant là où les lombricidés avaient disparu[112].
Une présence plus importante d'enchytréides (petits vers translucides habituellement associés aux sols asphyxiants, acides et pauvres) et de nématodes a été constatée dans les parcelles faiblement contaminées. La diversité des nématodes était plus grande à faible (0-5 cm) ou moyenne (5-10 cm) profondeur que dans les parcelles témoins ou celles très contaminées[112].
La proportion de « ravageurs » augmentait avec la contamination.
La respiration du sols diminuait sur les sols contaminés[112].
La contamination réduisait la nitrification, cependant la totalité de l'azote minéral était transformé en nitrates après 14 jours[112].
L'activité Sulfatase a été l'activité enzymatique la plus réduite par les fortes contaminations. À 100 mg/kg, le Cu, Cr et As n'ont causé qu'une faible inhibition de l'activité biologique globale du sol. Des effets plus délétères sur la vie du sol étaient observés à 400 et 800 mg/kg[112]. Dans le cas présent (où les apports en métaux ont cessé), les effets globaux sur la production fourragère ont semblé ne pas justifier une dépollution des sites[112]. - Dans des conditions anoxiques et réductrices (remontée de nappe ou inondation notamment), de l'arsenic pur peut être libéré des formes complexées ou organiques[113]. Dans les rizières il semble pouvoir être un facteur de stérilité du riz[114]
Synergies
Elles sont mal connues, mais elles existent.
- Par exemple, une exposition à l'arsenic seul ne semble pas avoir d'effet mutagène sur l'organisme ; mais si elle est associée à des radiations UV de courte longueur d'onde, l'arsenic agit comme un promoteur de tumeur dans le processus de carcinogénèse[115].
Cinétique environnementale
La cinétique de l'arsenic dans l'environnement varie selon le contexte pédogéologique, l'acidité des eaux lixiviantes, et sa spéciation.
Habituellement, l’arsenic est dispersé et se retrouve en faible quantité dans l’environnement. Les concentrations habituelles sont :
- dans les sols : entre 2 et 15 mg/L ;
- dans l’eau : moins de 10 µg/L ;
- dans les aliments : moins de 0,1 mg/kg ;
- dans l’air : entre 0,005 et 0,1 mg/m3.
Les activités minières (extraction du plomb, zinc argent mais aussi du fer[116] sont des sources de pollution par l'arsenic), la lixiviation et le ruissellement[40] sont contrôlés par de nombreux facteurs.
À titre d'exemple[40], en mésocosme de laboratoire de 0,9 m3, incluant 200 litres d'eau, après quatre mois de lixiviation d'un sol pollué par un pesticide arsenical, régulièrement arrosé, 0,6 % de l'arsenic total du sol (40 mg) a été relargué dans l'eau. Sur cette quantité relarguée, 7,5 % étaient en solution dans la colonne d'eau, 44 % étaient dans les sédiments peu profonds et 48,5 % plus profondément enfouis dans le sédiment. Dans la nature, le courant, la bioturbation et la bioconcentration peuvent modifier la cinétique de cet arsenic lessivé. Ici, différentes formes d'arsenic ; arsenite [As (III)], arséniate de [As (V)], acide monométhylarsonique (AMMA) et acide diméthylarsinique (DMAA), ont été recherchés dans l'eau lixiviée et dans l'eau interstitielle des sédiments[40]. Seul l'arséniate a été lessivé dans le sol. Dans l'eau du mésocosme il était l'espèce dissoute prédominante, mais du DMAA et des espèces particulaires ont aussi été détectés. Dans les sédiments superficiels, l'arséniate était ici encore l'espèce la plus abondante avec un peu de DMAA, alors que dans les sédiments profonds l'arsénite était prédominant[40].
Golfs
Les composés organo-arséniés actuellement utilisés dans les golfs ne sont pas réputés très toxiques pour l’être humain ou les animaux à sang chaud, mais leur décomposition dans l'environnement laisse des sous-produits arsenicaux inorganiques hautement toxiques, et éventuellement susceptibles de bioaccumulation ou bioconcentration.
L'association golfique USGA a sponsorisé une étude visant à modéliser le devenir dans le sol et l'eau d’infiltration des différentes formes (« espèces ») d'arsenic provenant du méthanearséniate monosodique (MSMA) notamment dans la zone racinaire du gazon. Pour cela un équivalent gazon de golf a été semé et entretenu par l’Université de Floride[117] et des lysimètres ont permis d'y mesurer la quantité d’eau percolant au travers des racines du gazon et dans le sol, ainsi que la quantité de nitrate et de MSMA lessivés au passage (avec évaluation de l'arsenic total et de sa spéciation). Il était intéressant d'évaluer les flux de nitrates en même temps que ceux d'arsenic, car l'arsenic (sous forme d'arsenite par exemple) agit synergiquement avec l’azote dans certains processus toxiques et d’acidification[118] Les résultats ont confirmé que comme dans les sols naturels ou agricoles, la composition du substrat (proportion de tourbe, argile et sable) influence fortement la mobilité et la lixiviation de l’arsenic[40], différemment selon ses espèces. Quatre produits de dégradation du MSMA étaient attendus. Ils ont été recherchés et trouvés dans les lysimètres ; l'arsénite (AsIII), l'arséniate (ASV), l'acide monométhylarsinique (MMAA) et l’acide diméthylarsinique (DMAA).
Vignes
Le métier de viticulteur est l'un de ceux qui étaient encore récemment les plus exposés aux pesticides arsénicaux. Ils ont été utilisés durant plus d'un siècle sur la vignes comme insecticides et fongicides. Théoriquement interdit en 1973, leur usage s'est en France prolongé grâce à une dérogation autorisant les vignerons à continuer à les utiliser (jusqu’en 2001) pour traiter l’Esca, une maladie fongique incurable du bois des ceps de vigne). Ces pesticides peuvent induire plusieurs types de cancers (carcinome basocellulaire cutané, carcinome spinocellulaire, cancer bronchique primitif, cancer des voies urinaires, adénocarcinome hépatocellulaire et angiosarcome du foie). De même que d'autres groupes de pesticides utilisés en agriculture, ils sont aussi suspectés d’augmenter le risque de maladie de Parkinson et de lymphome non hodgkinien[119], d'être des perturbateurs endocriniens et facteurs de délétion de la spermatogenèse. En 2018, une étude[119] a évalué qu'en vingt ans (de 1979 à 2000), le nombre d’ouvriers agricoles exposés a diminué de près de 40 % (passant de 101 359 à 61 376) alors que les effectifs des exploitations viticoles chutaient de plus de moitié en France, et alors que la « main d’œuvre familiale » de l’ensemble des exploitations agricoles de France métropolitaine restait stable (3,6 à 4,2 %) sur la période. L’exposition a augmenté parmi la main-d’œuvre familiale et la main-d’œuvre salariée des viticulteurs à but récréatif ou professionnel (10,5 à 19,6 %) et chez celles cultivant la vigne dans un but exclusivement professionnel (20 à 25 %). Les auteurs de l'étude estiment qu'un meilleur suivi post-professionnel est nécessaire, avec « le cas échéant, une reconnaissance en maladie professionnelle »[119].
Risques alimentaires
Il peut nous affecter via une contamination des aliments, ou via la pollution de nappes phréatiques, constatée notamment localement en Amérique du Nord, ou massivement en Asie du Sud-Est dans de grandes zones de culture du riz (Ouest du Bengale, Bangladesh et Viet-nam où au moins dix millions de personnes sont exposées à des taux d'arsenic dans l'eau dépassant les seuils de toxicité[120].) ; jusqu'à un million de puits creusés dans les dépôts alluviaux du Gange pourraient être contaminés par l'arsenic, avec des taux atteignant localement 1 000 mg/L au Bangladesh et au Bengale occidental et 3 000 µg/L au Viêt Nam ; l'irrigation du riz par une eau contaminée peut le contaminer[121] - [122] - [123] - [124] - [125].
Un Japonais moyen en 2006-2010 ingérait quotidiennement 2,31 μg/kg de poids corporel et par jour d'arsenic total (TA) ; et 0,260 μg/kg et par jour d'arsenic inorganique (iAs) [à comparer à 0,092 8 μg/kg et par jour de plomb, un métal souvent associé à l'arsenic dans la nature et l'industrie métallurgique], soit respectivement 138 μg d'arsenic total/personne et par jour, et 15,3 μg d'arsenic inorganique/personne et par jour, l'apport journalier variant selon le sexe, principalement en raison de la quantité d'aliments ingérés et beaucoup selon l'âge, le lieu de vie et le type de communauté (urbaine / agricole / de pêcheurs), « reflétant probablement la consommation de poisson/algues »[126].
Dans le monde (y compris en Europe) une source fréquente d'arsenic alimentaire est le riz (s'il est consommé abondamment et s'il provient de rizières naturellement riches en arsenic). En , l’Administration vétérinaire et alimentaire du Danemark (DVFA) a ainsi recommandé de ne pas donner trop de produits à base de riz aux enfants, en raison de leur teneur élevée en arsenic[127], de manière à ne pas dépasser les doses jugées dangereuses pour la santé par l'autorité européenne de sécurité des aliments (Efsa) qui a en 2009 modifié son évaluation de dose hebdomadaire tolérable (DHT) la portant à 15 µg/kg de poids corporel dose à partir de laquelle de premiers effets apparaissent pour la santé[128].
Normes
L'arsenic est un oligoélément, mais à très faible dose.
L’OMS, l’EPA (Environmental Protection Agency), Santé Canada, la France (en 2003) ou encore l’UE ont fixé la limite de concentration maximum de l’arsenic dans l’eau à 0,01 mg/L (10 µg/L[129]) tandis que le règlement sur la qualité de l’eau potable du Québec datant de 2001 fixait la limite à 0,025 mg/L. Cette limite a été diminuée en 2013 à 0,01 mg/L (10 µg/L[129]). Le Bangladesh et l’Inde où certaines régions sont naturellement, de par leur sous-sol et leurs nappes contaminées en arsenic fixent la limite à 0,05 mg/L.
Cette limite ne s'applique pas en France aux eaux minérales[130] mais uniquement à l'eau du robinet et aux eaux de source, de fait l'information sur la quantité d'arsenic n'apparait pas non plus sur l'étiquette informative des bouteilles, afin de ne pas porter préjudice à certaines marques inscrites dans le patrimoine national ou local.
Malgré ces normes, certains pays sont encore aujourd’hui souvent au-dessus des limites d’exposition selon l’OMS. Parmi eux, l’Argentine, l’Australie, le Bangladesh, le Chili, la Chine, les États-Unis, la Hongrie, l’Inde, le Mexique, le Pérou et la Thaïlande. Des effets négatifs sur la santé ont été observés au Bangladesh, en Chine, aux États-Unis et en Inde.
Des biomarqueurs peuvent avoir une valeur seuil spéciale, dédiée, dite « valeur de biomonitoring équivalent » (BE), quand elle sera convertie en doses externes à partir de modèles pharmacocinétiques, correspondront à une valeur de référence sanitaire établie ; il existe ainsi des systèmes de conversion (en quelque sorte en équivalent toxique) de l'arsenic organique vers l'arsenic minéral[131].
Recherche et développement
- 2007, France. Un groupe de chercheurs a mis en évidence que la bactérie Herminiimonas arsenicoxydans était capable, non seulement de transformer l’arsenic en une forme moins toxique, mais aussi capable d’isoler cet arsenic dans une matrice de sucres. (Pour rappel, au Bangladesh et en Chine, la concentration en arsenic dans l’eau dépasse les taux recommandés par l’OMS fixés à 0,01 mg/L).
- 2007, France et États-Unis. Par analogie avec le traitement d'une forme de la leucémie aiguë promyélocytaire, où il induit la dégradation d'une oncoprotéine spécifique[132], le trioxyde d'arsenic As2O3 a été utilisé avec succès sur un modèle animal de lupus érythémateux. Des chercheurs pensent pouvoir étendre son application à d'autres maladies auto-immunes[133].
- 2008 : Thomas Kulp et ses collègues de l'Institut d'études géologiques des États-Unis ont trouvé une nouvelle et unique forme de photosynthèse basée sur l’arsenic. Une « bactérie pourpre » et une cyanobactérie découvertes dans le Lac Mono (lac salé) en Californie peuvent vivre sans consommer d’eau libre, en oxydant l'arsénite (forme dissoute d’arsenic) pour en faire de l'arséniate, et ensuite fabriquer des molécules organiques. Une colonie de ces bactéries a pu être cultivée uniquement en présence d’arsénite. Il est possible que des bactéries de ce type ait fait partie des premières bactéries à peupler la Terre primitive[134]. Cette bactérie pourrait être bioindicatrice de sols très pollués par de l'arsenic.
- 2010 : La NASA découvre une bactérie dans le lac Mono capable de remplacer une des briques élémentaires de la vie, le phosphore, par l'arsenic. Cette annonce a depuis fait l'objet d'une réfutation[135].
Notes et références
- (en) Beatriz Cordero, Verónica Gómez, Ana E. Platero-Prats, Marc Revés, Jorge Echeverría, Eduard Cremades, Flavia Barragán et Santiago Alvarez, « Covalent radii revisited », Dalton Transactions, , p. 2832 - 2838 (DOI 10.1039/b801115j)
- (en) David R. Lide, CRC Handbook of Chemistry and Physics, CRC Press Inc, , 90e éd., 2804 p., Relié (ISBN 978-1-420-09084-0)
- Entrée « Arsenic » dans la base de données de produits chimiques GESTIS de la IFA (organisme allemand responsable de la sécurité et de la santé au travail) (allemand, anglais), accès le 22 août 2018 (JavaScript nécessaire)
- « Arsenic, elemental », sur Hazardous Substances Data Bank (consulté le )
- Base de données Chemical Abstracts interrogée via SciFinder Web le 15 décembre 2009 (résultats de la recherche)
- « Arsenic » dans la base de données de produits chimiques Reptox de la CSST (organisme québécois responsable de la sécurité et de la santé au travail), consulté le 25 avril 2009
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, « Evaluations Globales de la Cancérogénicité pour l'Homme, Groupe 1 : Cancérogènes pour l'homme », sur http://monographs.iarc.fr, CIRC, (consulté le )
- Directive 2004/107/CE du Parlement européen et du Conseil du 15 décembre 2004 concernant l’arsenic, le cadmium, le mercure, le nickel et les hydrocarbures aromatiques polycycliques dans l’air ambiant, Journal officiel de l'Union européenne, L23, 26 janvier 2005, p. 3-16.
- « L’arsenic, un nouveau type de perturbateur endocrinien ? », sur Caducee.net (consulté le ).
- Alain Foucault, op. cit.
- Source : Rapport Ifremer ; l’arsenic en milieu marin [PDF], étude de soutien à la définition de normes, Ifremer.
- (en) N. A. Gokcen, « The As (arsenic) system », Bulletin of Alloy Phase Diagrams, vol. 10, , p. 11–22 (DOI 10.1007/BF02882166, lire en ligne
 [PDF]).
[PDF]). - Hugues de Thé, « Differentiation Therapy Revisited », Nature Reviews Cancer, 18, no 2 (1er décembre 2017), 117‑27, DOI 10.1038/nrc.2017.103.
- Science et Vie, no 1075, avril 2007, p. 90.
- Lewis R. Goldfrank et Neal Flomenbaum, Goldfrank's toxicologic emergencies, chap. « Arsenicals ».
- Stéphane Gibaud et Gérard Jaouen, « Arsenic - based drugs: from Fowler’s solution to modern anticancer chemotherapy », Topics in Organometallic Chemistry, vol. 32, , p. 1-20 (DOI 10.1007/978-3-642-13185-1_1, lire en ligne)
- Catherine Quéré, Jean-Marie Sermier, Commission du développement durable Assemblée nationale (2015), Le vignoble français couvre environ 750 000 hectares répartis sur 25 000 communes. Le coût des maladies du bois, même si l’on manque d’un appareil statistique précis, est estimé à un milliard d’euros de manque à gagner, et son incidence économique et fiscale est indéniable, 7 juillet 2015.
- Agriculture-environnement, Le bourbier de l’arsénite de sodium, 24 mai 2005.
- Paul Depovere, La classification périodique des éléments. La merveille fondamentale de l'Univers, De Boeck Supérieur, , p. 98.
- Heather Lechtman, « Arsenic Bronze : Dirty Copper or Chosen Alloy ? A View from the Americas », Journal of Field Archaeology, vol. 23, no 4, , p. 477 (DOI 10.2307/530550)
- (en) Robert Jacobus Forbes, Studies in Ancient Technology : Metallurgy in Antiquity : Copper and Bronze, Tin, Arsenic, Antimony and Iron (Vol 9), Leiden, Brill Academic Pub, , 321 p. (ISBN 978-90-04-03487-7, BNF 37673394)
- Paul Benkimoun, « L'arsenic, un poison qui guérit », sur lemonde.fr,
- (en) Suzanne Bell, Barry A. J. Fisher et Robert C. Shaler, Encyclopedia of Forensic Science, Infobase Publishing, (lire en ligne), p. 219
- Albertus Magnus, De Mineralibus (le Monde minéral), Éditions du Cerf, , 443 p.
- Dietmar Seyferth, Cadet's Fuming Arsenical Liquid and the Cacodyl Compounds of Bunsen, Organometallics, 2001, vol. 20(8), p. 1488-1498, DOI 10.1021/om0101947.
- Le vert
- Jean-Antoine Chaptal, Chimie appliquée aux arts, Volume 4, chez Deterville, (lire en ligne), p. 467
- Fédération internationale du commerce des semences, Préparation des semences, Nyon, 2009.
- Thouin, H., Le Forestier, L., Gautret, P., Hube, D., Laperche, V., Dupraz, S. et Battaglia-Brunet, F. (2016), Characterization and mobility of arsenic and heavy metals in soils polluted by the destruction of arsenic-containing shells from the Great War. Science of the Total Environment, 550, 658-669.
- Extraits du décret du 14 septembre 1916 : Journal officiel du 20 septembre 1916, repris sur Pseudo-sciences.org.
- Lascar Olivier, « De la mort-aux-rats sur toiles », sur liberation.fr,
- C. Meyer, « Dictionnaire des Sciences Animales : Arsenic », Cirad, 18 avril 2012.
- 3-Nitro, produit par le laboratoire Pfizer subsidiary Alpharma LLC.
- C.W. Schmidt (2012), Maryland Bans Arsenical Drug in Chicken Feed, Environmental Health Perspectives, 120(7): a269 ncbi.nlm.nih.gov.
- Pfizer. Communiqué intitulé Pfizer to Suspend Sale of 3-Nitro (Roxarsone) in the United States, Madison, NJ:Pfizer Inc., 8 juin 2011, lire en ligne (consulté le 2012-06-07).
- Kawalek J.C. et al., Final Report on Study 275.30. Provide Data on Various Arsenic Species Present in Broilers Treated with Roxarsone: Comparison with Untreated Birds, Laurel, Rockville, and College Park, MD:Center for Veterinary Medicine/Office of Regulatory Science, U.S. Food and Drug Administration, 10 février 2011, lire en ligne (consulté le 7 juin 2012).
- Fisher D.J. et al. (2011), The Environmental Concerns of Arsenic Additives in Poultry Litter: Literature Review. Queenstown, MD:Wye Research and Education Center, , lire en ligne (consulté le 7 juin 2012).
- Arsenic Test Kits
- Turpeinen R, Pantsar-Kallio M, Haggblom M, Kairesalo T. 1999. Influence of microbes on the mobilization, toxicity and biomethylation of arsenic in soil. Science of the Total Environment 236: 173–180. (Résumé).
- M. Ruokolainen, M. Pantsar-Kallio, A. Haapa et T. Kairesalo, Leaching, runoff and speciation of arsenic in a laboratory mesocosm, Science of The Total Environment, vol. 258, no 3, 4 septembre 2000, p. 139-147 (Résumé).
- Van Herreweghe, S., Swennen, R., Vandecasteele, C. et Cappuyns, V. (2003), Solid phase speciation of arsenic by sequential extraction in standard reference materials and industrially contaminated soil samples, Environ. Pollut., 122 (3), 323–342. (Résumé).
- La Recherche, no 418, mars 2008, p. 10, en ligne : « Napoléon et la physique des neutrons ».
- Voir les fiches toxicologiques sur les sites de l’INRS et sur le site de REPTOX(Québec).
- Drobna Z, Jaspers I, Thomas DJ, Styblo M (2003), Differential activation of AP-1 in human bladder epithelial cells by inorganic and methylated arsenicals, The FASEB Journal, 17: 67–69. Lire en ligne.
- Caducee, d’après Nutrition et alimentation de B. Jacotot et J.-C. Le Parco.
- Autre estimation : 12 à 25 µg, Doctissimo, d’après Apports nutritionnels conseillés pour la population française, Agence française de sécurité sanitaire des aliments, 3e éd., Éd. Tec & Doc.
- Perez D.S., Armstrong-Lea L., Fox M.H., Yang R.S., Campain J.A. (), Arsenic and benzo[a]pyrene differentially alter the capacity for differentiation and growth properties of primary human epidermal keratinocytes, Toxicol Sci., décembre 2003, 76 (2), 280-90. Epub:2003-11-11.
- Salnikow K. et Cohen M.D. (2002), Backing into cancer: effects of arsenic on cell differentiation, Toxicol. Sci., fév 2002, 65(2):161-3 (résumé).
- : métaux et métalloïde des recherches de la cohorte Elfe ; décembre 2016 ; SANTÉ PUBLIQUE France / Imprégnation des femmes enceintes par les polluants de l’environnement en France en 2011. Volet périnatal du programme national de biosurveillance|PDF, 224p|Aussi disponible à partir de l’URL : www.santepubliquefrance.fr
- Dans ce cas, les auteurs précisent que seul l’arsenic total urinaire a été mesuré car le protocole de l’étude n’a pas permis de demander à ces femmes enceintes de ne pas manger de produits de la mer ou d’aliments connus pour être souvent anormalement contaminés au moins 72 h avant le prélèvement (qui a eu systématiquement lieu à l’arrivée à la maternité)
- INRS, Arsenic et composés inorganiques ; Nature du dosage : Arsenic urinaire, Base de données Biotox.
- Smith AH, Hopenhayn-Rich C, Bates MN, Goeden HM, Hertz-Picciotto I, Duggan HM, Wood R, Kosnett MJ, Smith MT. 1992. Cancer risk from arsenic in drinking water. Environmental Health Perspectives 103: 13–15. (Résumé).
- Yu HS, Liao WT, Chai CY (2006), Arsenic carcinogenesis in the skin ; J Biomed Sci. sept 2006 ; 13(5):657-66. Epub 2006 Jun 29 (résumé).
- Wang CH, Jeng JS, Yip PK, Chen CL, Hsu LI et al. (2002), Biological gradient between long-term arsenic exposure and carotid atherosclerosis. Circulation 105: 1804–1809
- Chen Y, Graziano JH, Parvez F et al., Arsenic exposure from drinking water and mortality from cardiovascular disease in Bangladesh: prospective cohort study, BMJ, 2011;362:d2431
- Von Ehrenstein OS, Mazumder DN, Yuan Y et al., Decrements in lung function related to arsenic in drinking water in West Bengal, India, Am J Epidemiol, 2005;162:533-41
- Liao WT, Yu CL, Lan CC, Lee CH, Chang CH, Chang LW, You HL, Yu HS (2009), Differential effects of arsenic on cutaneous and systemic immunity : focusing on CD4+ cell apoptosis in patients with arsenic-induced Bowen's disease ; Carcinogenesis, juin 2009 ; 30(6):1064-72. Epub 2009-04-17.
- J. Bastien et R. Jeandel, Napoléon à Sainte-Hélène. Étude critique de ses pathologies et des causes de son décès, Éd. Le Publieur, 2005, 220 p.
- Davey J.C., Bodwell J.E., Gosse J.A. et Hamilton J.W., Arsenic as an endocrine disruptor: effects of arsenic on estrogen receptor-mediated gene expression in vivo and in cell culture, Toxicol. Sci., juillet 2007, 98(1):75-86. Epub 2007-02-05 (résumé).
- Davey JC, Nomikos AP, Wungjiranirun M, Sherman JR, Ingram L, Batki C, Lariviere JP et Hamilton JW, Arsenic as an endocrine disruptor: arsenic disrupts retinoic acid receptor-and thyroid hormone receptor-mediated gene regulation and thyroid hormone-mediated amphibian tail metamorphosis, Environ. Health Perspect., février 2008, 116(2):165-72 (résumé).
- Bodwell JE, Kingsley LA, Hamilton JW (2004), Arsenic at very low concentrations alters glucocorticoid receptor (GR)-mediated gene activation but not GR-mediated gene repression: complex dose-response effects are closely correlated with levels of activated GR and require a functional GR DNA binding domain. ; Chem Res Toxicol. aout 2004 ; 17(8):1064-76 (résumé).
- Aquino A.M. et al. (2019), Arsenic exposure during prepuberty alters prostate maturation in pubescent rats. Reproductive Toxicology, 89, 136-144 (résumé).
- Ghosh M, Shen J, Rosen BP. 1999. Pathways of As (III) detoxification in Sachharomyces cerevisiae. Proceedings of the National Academy of Sciences, États-Unis 96: 5001–5006 (Résumé).
- Clemens S. 2001. Molecular mechanisms of plant metal tolerance and homeostasis, Planta, 212, 475–486 (Résumé)).
- Cobbett C.S. (2000), Phytochelatin biosynthesis and function in heavy-metal detoxification, Current Opinion in Plant Biology, 3, 211–216 (Résumé).
- Nickson R., McArthur J., Burgess W., Ahmed K.M., Ravenscroft P., Rahmann M. (1998), Arsenic poisoning of Bangladesh groundwater. Nature, 395: 338.
- Woolson EA, Axley JH, Kearney PC. (1971), The chemistry and phytotoxicity of arsenic in soils. I. Contaminated field soils, Soil Science Society of America Proceedings, 35: 938–943. (Résumé).
- Murphy E.A. et Aucott M. (1998), An assessment of the amounts of arsenical pesticides used historically in a geographical area, Science of the Total Environment, 218, 89-101 (Résumé).
- Cullen W.R. et Reimer K.J. (1989), Arsenic speciation in the environment, Chemical Review, 89, 713-764 (Résumé).
- Andrew A. Meharg, Jeanette Hartley-Whitaker (2002), Arsenic uptake and metabolism in arsenic resistant and nonresistant plant species ; en ligne le 4 avril 2002, DOI 10.1046/j.1469-8137.2002.00363.x, Issue New Phytologist, vol. 154, no 1, p. 29-43, avril 2002 (résumé).
- « Eawag (2015) Geogenic Contamination Handbook – Addressing Arsenic and Fluoride in Drinking Water. C.A. Johnson, A. Bretzler (Eds.), Swiss Federal Institute of Aquatic Science and Technology (Eawag), Duebendorf, Switzerland ».
- Amini, M. ; Mueller, K. ; Abbaspour, K.C. ; Rosenberg, T. ; Afyuni, M. ; Møller, M. ; Sarr, M. et Johnson, C.A., « Statistical modeling of global geogenic fluoride contamination in groundwaters », Environmental Science and Technology, 42(10), 3662-3668, (DOI 10.1021/es071958y).
- Amini, M. ; Abbaspour, K.C. ; Berg, M. ; Winkel, L. ; Hug, S.J. ; Hoehn, E. ; Yang, H. et Johnson, C.A., « Statistical modeling of global geogenic arsenic contamination in groundwater », Environmental Science and Technology 42 (10), 3669-3675, (DOI 10.1021/es702859e).
- Winkel, L. ; Berg, M. ; Amini, M. ; Hug, S.J. et Johnson, C.A., « Predicting groundwater arsenic contamination in Southeast Asia from surface parameters », Nature Geoscience, 1, 536–542 (2008), (DOI 10.1038/ngeo254)
- Rodríguez-Lado, L. ; Sun, G. ; Berg, M. ; Zhang, Q. ; Xue, H. ; Zheng, Q. et Johnson, C.A., « Groundwater arsenic contamination throughout China », Science, 341(6148), 866-868, (DOI 10.1126/science.1237484)
- Cheng H., Hu Y., Luo J., Xu B., Zhao J. J. Hazard Mater (2009), Geochemical processes controlling fate and transport of arsenic in acid mine drainage (AMD) and natural systems ; 15 juin 2009, 165(1-3):13-26. Epub 25 octobre 2008 (résumé).
- Nriagu J.O. 1994. Arsenic in the environment: part 1 cycling and characterization, New York, États-Unis, Wiley, 430.
- Min Feng, Jill E. Schrlau, Raymond Snyder, George H. Snyder, Ming Chen, John L. Cisar et Yong Ca, Arsenic Transport and Transformation Associated with MSMA Application on a Golf Course Green [PDF], J. Agric. Food Chem., 2005, 53 (9), p. 3556–3562, DOI 10.1021/jf047908j (Résumé).
- Voir graphique fait d'après données Medde, DGPR (Basol au 16 janvier 2012), Traitements : SOeS, 2012 ; p. 21, dans Commissariat général au développement durable, Basol : un panorama des sites et sols pollués, ou potentiellement pollués, nécessitant une action des pouvoirs publics, Études & documents, no 97, novembre 2013.
- Réflexions sut l'environnement, sur ecoumenegolf.org.
- (en) David Jordan, Marilyn McClelland, Andy Kendig, et Robert Frans, Monosodium Methanearsonate Influence on Broadleaf Weed Control with Selected Postemergence-Directed Cotton Herbicides [PDF], Weed Science, The Journal of Cotton Science, 1:72-75 (1997), 4 p.
- Bausinger Tobias ; Preuß Johannes, Environmental remnants of the First World War: Soil contamination of a burning ground for arsenical ammunition. Dans Bulletin of Environmental Contamination & Toxicology, 74, 2005, p. 1045-1052.
- T. Bausinger et al., « Exposure assessment of a burning ground for chemical ammunition on the Great War battlefields of Verdun », Science of the Total Environment, 382, 2007, p. 259-271.
- Camille Larue et Marie Carrière, Les nanoparticules dans l’écosystème sol ; Période : février 2010 à août 2010 CEA/ADEME – IRAMIS/SIS2M/LSDRM – Gif-sur-Yvette.
- Larios R., Fernández-Martínez R., Lehecho I. et Rucandio I. (2012), A methodological approach to evaluate arsenic speciation and bioaccumulation in different plant species from two highly polluted mining areas, Sci. Total Environ., 1er janvier 2012, 414:600-7, Epub 10 décembre 2011 (résumé).
- Larios R, Fernández-Martínez R, Silva V, Loredo J, Rucandio I (2012), Arsenic contamination and speciation in surrounding waters of three old cinnabar mines. J Environ Monit. 2012 Feb; 14(2):531-42. Epub 2011 Dec 5 (résumé).
- Sheppard, S.C. (1992), Summary of phytotoxic levels of soil, Water Air Soil Pollut., vol. 64, no 3-4, p. 539-550 ; septembre 1992 (ISSN 0049-6979) (Résumé, sur Web of science).
- Fitter AH, Wright WJ, Williamson L, Belshaw M, Fairclough J, Meharg AA. 1998. The phosphorus nutrition of wild plants and the paradox of arsenate tolerance: does leaf phosphate concentration control flowering ? In: LynchJP, DeikmanJ, eds. Phosphorus in plant biology: regulatory roles in molecular, cellular, organismic and ecosystem processes. American Society of Plant Physiologists, 39–51
- Francesconi K, Visoottiviseth P, Sridockhan W, Goessler W. 2002. Arsenic species in an hyperaccumulating fern, Pityrogramma calomelanos: a potential phytoremediator. Science of the Total Environment, 284: 27–35.
- Junru Wang, Fang-Jie Zhao, Andrew A. Meharg, Andrea Raab, Joerg Feldmann et Steve P. McGrath, Mechanisms of Arsenic Hyperaccumulation in Pteris vittata. Uptake Kinetics, Interactions with Phosphate, and Arsenic Speciation ; Plant Physiology, novembre 2002, vol. 130, no 3, 1552-1561, en ligne, octobre 2002, DOI 10.1104/pp.008185 ((en) Résumé).
- Andrea Raab, Jörg Feldmann et Andrew A. Meharg, Environmental stress and adaptation ; The Nature of Arsenic-Phytochelatin Complexes in Holcus lanatus and Pteris cretica ; Plant Physiology, 134:1113-1122 (2004) (American Society of Plant Biologists).
- Enzo Lombi, Fang-Jie Zhao, Mark Fuhrmann, Lena Q. Ma et Steve P. McGrath, Arsenic distribution and speciation in the fronds of the hyperaccumulator Pteris vittata, en ligne le 21 octobre 2002, DOI 10.1046/j.1469-8137.2002.00512.x.
- ma et al., Nature, 2001, 409:579.
- Porter, E.K. 1977. Arsenic tolerance in grasses growing on mine waste. Environ. Pollut., 14:255–265 (Résumé).
- De Koe T, Jacques NMM. 1993. Arsenate tolerance in Agrostis castellana and Agrostis delicatula. Plant and Soil 151: 185–191. (Résumé).
- Castillo-Michel H, Hernandez-Viezcas J, Dokken KM, Marcus MA, Peralta-Videa JR, Gardea-Torresdey JL (2011), Localization and speciation of arsenic in soil and desert plant Parkinsonia florida using μXRF and μXANES ; Environ Sci Technol. 2011 Sep 15;45(18):7848-54. DOI 10.1021/es200632s. Epub 2011 Aug 26. (résumé).
- Pitten, Frank-Albert, Müller Gerald, König Peter et al., Arsenakkumulation in Pflanzen, Abschlußbericht AP 120/2. Greifswand, 1997, unveröffentlicht, p. 60.
- Jiang, Q.Q. 1994. Effect of different forms and sources of arsenic on crop yield and arsenic concentration. Water Air Soil Pollut. 74:321–343. (Résumé).
- Frans R., Horton D. et Burdette L. (1988), Influence of MSMA on straighthead. Arsenic uptake and growth-response in rice (Oryza Sativa), Arkansas Agricultural Experiment Station Report Series 30: 1–12.
- Marin, A.R. (1992), The influence of chemical form and concentration of arsenic on rice growth and tissue arsenic concentration, Plant Soil, 139:175–183, DOI 10.1007/BF00009308.
- Carbonell-Barrachina, A.A. (1997), The influence of arsenite concentration on arsenic accumulation in tomato and bean plants, Sci. Hortic., 71, 167–176, DOI 10.1016/S0304-4238(97)00114-3.
- Edmonds JS. 2000. Diastereoisomers of an ‘arsenomethionine’-based structure from Sargassum lacerifolium: The formation of the arsenic-carbon bond in arsenic-containing natural products. Bioorganic and Medicinal Chemistry Letters 10: 1105–1108 (Résumé).
- Helgesen H. et Larsen E.H. (1998), Bioavailability and speciation of arsenic in carrots grown in contaminated soil, Analyst, 123: 791–796 (Résumé).
- Khan, Masil, Effect of metals contamination on soil microbial diversity, enzymatic activity, organic matter decomposition and nitrogen mineralization (a review) ; Pakistan Journal of Biological ; Sciences, 2000 - 198.170.104.138.
- C. Leyval, K. Turnau et K. Haselwandter (1997) Effect of heavy metal pollution on mycorrhizal colonization and function: physiological, ecological and applied aspects ; Biomedical and Life Sciences Mycorrhiza Volume 7, Number 3, 139-153, DOI 10.1007/s005720050174 (Résumé).
- Y. Liu, Y. G. Zhu, B. D. Chen, P. Christie et X. L. Li (2005), Original Paper Influence of the arbuscular mycorrhizal fungus Glomus mosseae on uptake of arsenate by the As hyperaccumulator fern Pteris vittata L., Biomedical and Life Sciences Mycorrhiza, vol. 15, no 3, 187-192, DOI 10.1007/s00572-004-0320-7 (Résumé).
- Gonzalez-Chavez M. 2000. Arbuscular mycorrhizal fungi from As/Cu polluted soils. PhD thesis. University of Reading, Royaume-Uni.
- C. Gonzalez-Chavez, P. J. Harris, J. Dodd, A. A. Meharg (2002), Arbuscular mycorrhizal fungi confer enhanced arsenate resistance on Holcus lanatus ; New Phytologist ; vol. 155, no 1, p. 163–171, juillet 2002 (Résumé).
- Wolfe-Simon et al., A bacterium that can grow by using arsenic instead of phosphorus, Science (2011), vol. 332 (6034) p. 1163-6.
- Reaves et al., Absence of Detectable Arsenate in DNA from Arsenate-Grown GFAJ-1 Cells, Science (2012).
- Vishnu Priya Gadepalle, Sabeha K. Ouki, René Van Herwijnen et Tony Hutchings ; Effects of amended compost on mobility and uptake of arsenic by rye grass in contaminated soil ; Chemosphere ; Volume 72, no 7, July 2008, p. 1056-1061, DOI 10.1016/j.chemosphere.2008.03.048 ((en) résumé).
- G. W. Yeates, V. A. Orchard, T. W. Speir, J. L. Hunt and M. C. C. Hermans, Impact of pasture contamination by copper, chromium, arsenic timber preservative on soil biological activity ; Biology and Fertility of Soils, vol. 18, no 3, 200-208, 1984, DOI 10.1007/BF00647667 (Résumé).
- Rochette, E.A. 1998. Stability of arsenate minerals in soil under biotically generated reducing conditions. Soil Sci. Soc. Am. J. (Home » Publications » Soil Science Society of America Journal) 62:1530–1537 (Résumé).
- Gilmour J, Wells B. 1980. Residual effects of MSMA on sterility in rice cultivars. Agronomy Journal 72: 1066–1067 (Résumé).
- Jung D.K. et al., Hydrogen peroxide mediates arsenite activation of p70(s6k) and extracellular signal-regulated kinase, Exp. Cell Res., 2003, 290 (1), 144-54.
- Courtin-Nomade et al. (2003), Arsenic in iron cements developed within tailings of a former metalliferous mine - Enguialès, Aveyron, France, Applied Geochemistry, 18, p. 395-408.
- Lieu : « Fort Lauderdale Research and Education Center » (FLREC)
- Gurr J.R. et al., Nitric oxide production by arsenite, Mutat. Res., 2003, 533 (1-2), 173-82.
- Spinosi J., Chaperon L., Jezewski-Serra D. et El Yamani M. (2018), Occupational exposure of winegrowers to arsenical pesticides: Exposure prevalence between 1979 and 2000, Santé publique France et l'Umrestte de l'université de Lyon.
- Christen K. (2001), The arsenic threat worsens, Environmental Science and Technology, 35: 286A–291A (Résumé).
- Nickson R, McArthur J, Burgess W, Ahmed KM, Ravenscroft P, Rahmann M. 1998. Arsenic poisoning of Bangladesh groundwater. Nature 395: 338.
- Berg M, Tran HC, Nguyen TC, Pham HV, Schertenleib R, Giger W. 2001. Arsenic contamination of groundwater and drinking water in Viêt Nam: a human health threat. Environmental Science and Technology 35: 2621–2626 (Résumé).
- Christen K. 2001. The arsenic threat worsens. Environmental Science and Technology 35: 286A–291A. (Résumé).
- Abedin J, Cresser M, Meharg AA, Feldmann J, Cotter-Howells J. 2002. Arsenic accumulation and metabolism in rice (Oryza sativa L.). Environmental Science and Technology.
- Abedin MJ, Feldmann J, Meharg AA. 2002. Uptake kinetics of arsenic species in rice (Oryza sativa L.) plants. Plant Physiology.
- Hayashi, A., Sato, F., Imai, T. et Yoshinaga, J. (2019), Daily intake of total and inorganic arsenic, lead, and aluminum of the Japanese: Duplicate diet study, Journal of Food Composition and Analysis, 77, 77-83 (résumé).
- Romain Loury (2013), Moins de riz pour les petits Danois 2013-05-22
- (en) « EFSA - Scientific Opinion of the CONTAM Panel: Arsenic in Food », sur EFSA.
- EPA, EPA 815-R-00-026 Arsenic in Drinking Water Rule Economic Analysis
- Rapport sur la qualité de l'eau et l'assainissement en France (www.senat.fr)
- Hays S.M., Aylward L.L., Gagne M., Nong A. et Krishnan K., Biomonitoring equivalents for inorganic arsenic, RegulToxicolPharmacol., 2010, 58 (1), 1-9.
- Hugues de Thé, « Pourquoi l'arsenic est-il si efficace dans le traitement d'une leucémie ? », sur cnrs.fr, (consulté le )
- revue Blood, vol.108, no 13, p. 3967-3975
- Brève scientifique de Pour la Science, no 372, octobre 2008 (p. 14, citant Science, vol. 321, p. 967-970, août 2008).
- Reaves ML, Sinha S, Rabinowitz JD, Kruglyak L et Redfield RJ, Absence of detectable arsenate in DNA from arsenate-grown GFAJ-1 cells, Science, 2012;337:470-3.
Voir aussi
Bibliographie
- Centre d'expertise en analyse environnementale du Québec, Détermination de l’arsenic dans l’eau ; Méthode automatisée par spectrophotométrie d’absorption atomique et formation d’hydrures. MA. 203 – As 1.0, ministère de l’Environnement du Québec, 2003, 17 p.
- Centre d'expertise en analyse environnementale du Québec, Détermination de l’arsenic dans les sédiments : méthode automatisée par spectrophotométrie d’absorption atomique après minéralisation et génération d’hydrure. MA. 205 – As 1.0, ministère de l’Environnement du Québec, 2003, 17 p.
- Commission sur la gestion de l’eau au Québec, Rapport no 142, L’eau, ressource à protéger, à partager et à mettre en valeur, t. II, Bureau d’audiences publiques sur l’environnement.
- (en) EPA, Method 200.8, Determination of trace elements in water and wastes, by inductively coupled plasma-mass spectrometry, Revision 5.4, EMMC Version.
- (en) EPA (Office of prevention, pesticide and toxic substance) « Revised Reregistration Eligibility Decision for MSMA, DSMA, CAMA, and Cacodylic Acid »(Archive.org • Wikiwix • Archive.is • Google • Que faire ?) [PDF], (consulté le ), Mémorandum, 70 p.
- (fr)/(en), Alain Foucault, Jean-François Raoult, Fabrizio Cecca et Bernard Platevoet, Dictionnaire de Géologie, 8e éd., Dunod, 2014, 416 p. Avec la simple entrée « arsenic », p. 26-27.
- Groupe scientifique sur l’eau (2002), « Arsenic », Dans Fiches synthèse sur l’eau potable et la santé humaine, Institut national de santé publique du Québec, 8 p.
- Laperche V., Bodénan F., Dictor M.C. et Baranger Ph. (2003), Guide méthodologique de l’arsenic, appliqué à la gestion des sites et sols pollués, BRGM/RP-52066-FR, 90 p., 5 fig., 10 tabl., 3 ann.
- Liao W.T., Lan C.C., Lee C.H. et Yu H.S., Concentration-dependent cellular responses of arsenic in keratinocytes ; Kaohsiung J. Med. Sci., septembre 2011 ; 27(9):390-5, Epub 2011-08-09.
- OMS, Aide-mémoire no 210, révisé mai 2001, L’arsenic dans l’eau de boisson.
- Jean Perrotey, « arsenic », Encyclopædia Universalis, 2001, début en ligne.
- Québec ; Règlement sur la qualité de l’eau potable, Québec.
- (en) Gong Z. et al., Arsenic speciation analysis, Talanta, 58 (2002) 77–96.
- Santé Canada, Méthodes d’analyses - Arsenic - Recommandations pour la qualité de l’eau potable au Canada : document technique.
- Louis Troost, Traité élémentaire de chimie, 6e éd., Paris, 1880, en particulier l'« arsenic », p. 191, en ligne, 12e éd.
- Ullrich-Eberius C.I., Sanz A. et Novacky A.J. (1989), Evaluation of arsenate- and vanadate-associated changes of electrical membrane potential and phosphate transport in Lemna gibba G1. Journal of Experimental Botany, 40: 119–128 (Résumé).
- Visoottiviseth P, Francesconi K, Sridokchan W. (2002), The potential of Thai indigenous plant species for the phytoremediation of arsenic contaminated land. Environmental Pollution 118: 453–461 (Résumé).
- Webb SM, Gaillard JF, Ma LQ, Tu C. 2001, Arsenic speciation in a hyper-accumulating fern using CS-XAS. American Chemical Society, Environmental Chemistry Extended Abstracts 222: 60-62.
- Zhao FJ, Dunham SJ, McGrath SP. (2002), Arsenic hyperaccumulation by different fern species. New Phytologist 156: (Résumé).
Bases de données toxicologiques
- INERIS, Fiche de données toxicologiques et environnementales des substances chimiques, Arsenic et ses dérivés inorganiques, Version no 3, juillet 2006.
- INRS, Arsenic et composés inorganiques (dosés dans l'urine / Arsenic urinaire) ; Base de données Médecine du travail / toxicologie « Biotox ».
- (en) ATSDR, Toxic Substances Portal–Arsenic : Public Health Statement for Arsenic, Atlanta, GA, Agency for Toxic Substances & Disease Registry, U.S. Centers for Disease Control and Prevention.
Dans la fiction
- Madame Bovary s'empoisonne à l'arsenic.
- Le « pudding à l'arsenic » empoisonne Cléopâtre dans Astérix et Cléopâtre.
- Arsenic et vieilles dentelles est une pièce de théâtre américaine.
Articles connexes
- Acétarsol
- Acide arsanilique
- Acide arsénique
- Acide arsonique
- Acide cacodylique
- Acide métaarsénieux
- Acide pyroarsénique
- Acide orthoarsénieux
- Adamsite (arme chimique)
- Agent bleu
- anhydride arsénieux ou trioxyde d'arsenic
- Arsanilate de sodium
- Arséniate
- Arséniate de gallium
- Arséniate de potassium
- arséniate de plomb
- Arsenic (maladie professionnelle)
- Arsenic cancers (maladie professionnelle)
- Arsenic natif
- Arsénite
- Arsénite de cuivre(II)
- Arsénite méthyltransférase
- Arséniure d'aluminium
- Arsénobétaïne
- Arsénolamprite
- Arsénopyrite
- Arsphénamine
- Arsthinol
- Bronze arsénié
- Cacodyle
- Composé organo-arsénié
- Écotoxicologie
- Herminiimonas arsenicoxydans
- Hydrogène arsénié (maladie professionnelle)
- Hydrogénoarsénite de cuivre
- Intoxication par l'arsenic
- Isotopes de l'arsenic
- Mélarsoprol
- Métaarsénite de cuivre(II)
- Métaarsénite de sodium
- Méthanearséniate monosodique
- Méthylarsonate réductase
- Mort aux rats
- Orpiment
- Orpiment (pigment)
- Oxyde d'arsenic
- Oxyde de cacodyle
- Pentoxyde d'arsenic
- Phospho-arséniure de gallium
- Pnictogène
- Poison
- Réalgar
- Séquelle de guerre
- Stibarsen
- Test de Gutzeit
- Toxicologie
- Trihydrure d'arsenic
- Triméthylarsine
Liens externes
- Présentation succincte de l'élément, sur lenntech.fr
- Présentation de l'élément, sur futura-sciences.com
- (en) Images de l'arsenic, sous différentes formes
- Arsenic et santé, résumé de GreenFacts d’un rapport scientifique du Programme International sur la Sécurité Chimique de l’OMS
- Étude de l’INVS (10/09/08) sur l’exposition au plomb, cadmium et à l’arsenic par des sols pollués en Aveyron.
- Document canadien sur l’arsenic
- Arsenic en milieu marin [PDF], 65 p., Ifremer, 1993
- (en) Marsh Test en visuel [vidéo], sur YouTube
- (en) Conférence sur la pollution des eaux et aquifères par As [vidéo], sur YouTube
- (en) « Technical data for Arsenic » (consulté le ), avec en sous-pages les données connues pour chaque isotope
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | ||||||||||||||||
| 1 | H | He | |||||||||||||||||||||||||||||||
| 2 | Li | Be | B | C | N | O | F | Ne | |||||||||||||||||||||||||
| 3 | Na | Mg | Al | Si | P | S | Cl | Ar | |||||||||||||||||||||||||
| 4 | K | Ca | Sc | Ti | V | Cr | Mn | Fe | Co | Ni | Cu | Zn | Ga | Ge | As | Se | Br | Kr | |||||||||||||||
| 5 | Rb | Sr | Y | Zr | Nb | Mo | Tc | Ru | Rh | Pd | Ag | Cd | In | Sn | Sb | Te | I | Xe | |||||||||||||||
| 6 | Cs | Ba | La | Ce | Pr | Nd | Pm | Sm | Eu | Gd | Tb | Dy | Ho | Er | Tm | Yb | Lu | Hf | Ta | W | Re | Os | Ir | Pt | Au | Hg | Tl | Pb | Bi | Po | At | Rn | |
| 7 | Fr | Ra | Ac | Th | Pa | U | Np | Pu | Am | Cm | Bk | Cf | Es | Fm | Md | No | Lr | Rf | Db | Sg | Bh | Hs | Mt | Ds | Rg | Cn | Nh | Fl | Mc | Lv | Ts | Og | |
| 8 | 119 | 120 | * | ||||||||||||||||||||||||||||||
| * | 121 | 122 | 123 | 124 | 125 | 126 | 127 | 128 | 129 | 130 | 131 | 132 | 133 | 134 | 135 | 136 | 137 | 138 | 139 | 140 | 141 | 142 | |||||||||||