Paracétamol
Le paracétamol, aussi appelé acétaminophène, est un composé chimique utilisé comme antalgique (anti-douleur) et antipyrétique (anti-fièvre), qui figure parmi les médicaments les plus communs, utilisés et prescrits au monde.
| Paracétamol | ||
| ||
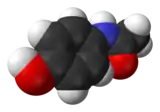 | ||
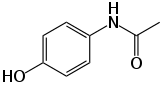
| Représentations plane et 3D d'une molécule de paracétamol | ||
| Identification | ||
|---|---|---|
| DCI | Paracétamol[1] | |
| Nom UICPA | N-(4-hydroxyphényl)acétamide | |
| Synonymes |
acétaminophène |
|
| No CAS | ||
| No ECHA | 100.002.870 | |
| No CE | 203-157-5 | |
| No RTECS | AE4200000 | |
| Code ATC | N02 | |
| DrugBank | DB00316 | |
| PubChem | 1983 | |
| ChEBI | 46195 | |
| SMILES | ||
| InChI | ||
| Apparence | poudre blanche cristallisée inodore[2] | |
| Propriétés chimiques | ||
| Formule | C8H9NO2 [Isomères] |
|
| Masse molaire[3] | 151,162 6 ± 0,007 8 g/mol C 63,56 %, H 6 %, N 9,27 %, O 21,17 %, |
|
| pKa | 9,38[4] | |
| Propriétés physiques | ||
| T° fusion | 169 à 171 °C[2] | |
| T° ébullition | décomposition | |
| Solubilité | 14 g L−1[2] à 20 °C ; bien plus soluble dans l'eau chaude. Soluble dans l'acétone, l'éthanol, le méthanol, le diméthylformamide, le dichlorure d'éthylène, l'acétate d'éthyle. Peu soluble dans le chloroforme, l'éther. Presque insoluble dans l'éther de pétrole, le pentane, le benzène. |
|
| Masse volumique | 1,293 g cm−3[2] à 21 °C | |
| T° d'auto-inflammation | 540 °C[2] (inflammation brève sans propagation) | |
| Point d’éclair | 177 °C[5] | |
| Cristallographie | ||
| Classe cristalline ou groupe d’espace | P21/n[6] | |
| Paramètres de maille | a = 7,094 Å b = 9,232 Å |
|
| Volume | 753,94 Å3[6] | |
| Précautions | ||
| SGH[2] | ||
 Attention |
||
| SIMDUT[7] | ||
Produit non contrôlé |
||
| Classification du CIRC | ||
| Groupe 3 : Inclassable quant à sa cancérogénicité pour l'Homme[8] | ||
| Écotoxicologie | ||
| DL50 | 1 940 mg/kg[2] (souris, oral) 800 mg/kg souris i.p. 825 mg/kg chien i.v. |
|
| LogP | 0,49[9] | |
| Données pharmacocinétiques | ||
| Biodisponibilité | Proche de 100 % | |
| Métabolisme | Hépatique à 95 % | |
| Demi-vie d’élim. | 1 à 4 heures | |
| Excrétion | ||
| Considérations thérapeutiques | ||
| Classe thérapeutique | Antalgique • Antipyrétique | |
| Voie d’administration | Orale, IV, intrarectale | |
| Grossesse | Autorisé | |
| Précautions | Toxicité hépatique à fortes doses | |
| Antidote | N-acétylcystéine | |
| Unités du SI et CNTP, sauf indication contraire. | ||
Il est indiqué dans le traitement des douleurs d'intensité faible à modérée, seul ou en association avec d'autres analgésiques opioïdes. Il est très populaire car il a moins de contre-indications que les autres antalgiques et jouit d'une bonne image auprès du public.
Son mécanisme est très complexe ; en effet, il diminue la fièvre, mais pas par le même mécanisme que l'aspirine ou l'ibuprofène, qui agissent sur l'inflammation[alpha 1] - [10].
Le paracétamol est le médicament le plus prescrit en France, et même la base des trois médicaments les plus prescrits (noms commerciaux : Doliprane, Dafalgan, Efferalgan), qui totalisent plus de 500 millions de boîtes en 2013[11].
Toutefois, au-delà de 6 g/j, et en cas d'usage au long cours, le paracétamol présente une sévère toxicité pour le foie par production d'un métabolite hépatotoxique, la N-acétyl-p-benzoquinone imine (NAPQI). En cas de surdose ou d'interactions médicamenteuses, le paracétamol est très toxique pour le foie et peut entraîner la mort par hépatite fulminante. C'est pourquoi il est fortement contre-indiqué chez les personnes souffrant d'insuffisance hépatique. La N-acétylcystéïne est un précurseur du glutathion qui diminue la toxicité du paracétamol en inactivant le métabolite toxique du paracétamol NAPQI (N-acétyle-p-benzoquinone imine). Cet antidote est plus efficace s'il est administré dans les huit heures qui suivent l'ingestion de paracétamol. Après 24 heures, le bénéfice de l'antidote est discutable, mais il doit cependant être administré. Si le degré de toxicité est incertain, de la N-acétylcystéïne doit être administrée jusqu'à ce qu'une intoxication soit exclue. Une dose de charge de 150 mg/kg en 200 mL de glucosé à 5 % administrée en 15 min est suivie par des doses d'entretien de 50 mg/kg dans 500 mL de glucosé à 5 % administrés en quatre heures, puis 100 mg/kg dans 1 000 mL de glucosé à 5 % administrés en seize heures. Chez l'enfant, il faut parfois ajuster le dosage pour diminuer le volume total de liquide qui est administré ; une consultation dans un centre antipoison est recommandée[12].
Le nom « paracétamol » vient de la contraction de para-acétyl-amino-phénol. Acétaminophène quant à lui provient de N-acétyl-para-aminophénol. Il est appelé acétaminophène dans les pays utilisant la dénomination USAN (United States Adopted Name) : au Canada, aux États-Unis, au Mexique, en Colombie, au Japon, en Corée du Sud, à Hong Kong et en Iran. Dans les pays utilisant la dénomination commune internationale, on emploie le nom « paracétamol ».
Historique
L'usage d'antipyrétiques remonte à l'Antiquité : ils étaient des préparations à partir de composés naturels d'écorces de cinchona[13] (dont dérive la quinine), soit du salicylate (dont dérive l'aspirine) contenu dans l'écorce de saule. Les substances de synthèse sont fabriquées en laboratoire, et non plus extraites directement de substances naturellement présentes.
Harmon Northrop Morse (en) synthétisa dès 1878 une substance baptisée acétylaminophénol[14], sans toutefois lui attribuer une quelconque propriété médicale. C'est cinquante ans plus tard qu'elle fut commercialisée comme médicament sous le nom de paracétamol. À cette époque, d'autres produits sont utilisés comme remède contre la douleur et la fièvre : en 1882, Hoechst commercialise le Kairin découvert par Otto Fisher[15] ; en 1897, l'aspirine est synthétisée par Felix Hoffmann et connaît un grand succès. BASF ne pousse guère son antipyrétique Thallin, mis au point vers 1885. L'acétanilide (1886) et la phénacétine (1887) sont également utilisées avant qu'on ne constate les graves effets secondaires de leur administration, tandis que les inconvénients de l'aspirine commencent à être connus. Le paracétamol réapparaît alors et les premières études sur les propriétés antipyrétiques et antalgiques du paracétamol sont conduites à la fin du XIXe siècle.
En 1886, le professeur Adolf Kussmaul, de l'université de Strasbourg, étudie l'effet antiparasitaire du naphtalène. Ses deux jeunes assistants Arnold Cahn et Paul Hepp, à court de produit pour leurs expériences, décident de se ravitailler auprès d'un pharmacien de la ville, qui leur fournit par erreur de l'acétanilide. En reprenant leur étude, ils sont intrigués par les effets antipyrétiques qu'ils obtiennent avec lui. C'est donc grâce à une erreur providentielle — autrement dit par sérendipité — que les propriétés de l'acétanilide contre la fièvre sont découvertes[16]. En 1861, Schuchardt avait déjà découvert les propriétés antipyrétiques de l'aniline, substance servant à la production d'acétanilide (du fait d'effets toxiques prononcés, l'aniline n'eut cependant pas d'utilisation en thérapeutique) ; ses propriétés antalgiques seront découvertes un peu plus tard. L'acétanilide est l'ancêtre du paracétamol et de la phénacétine[17]. Le docteur Hepp a un frère travaillant pour une petite compagnie (Kalle Co) qui fabrique l'acétanilide[18]. Il lui propose d'utiliser sa découverte et de lancer sur le marché l'acétanilide afin de concurrencer l'antipyrine[19] - [20] et l'acide salicylique. L'acétanilide devient un médicament commercialisé sous le nom d'antifébrine[21].
À la fin des années 1880, l'industrie des colorants avait un déchet, le paranitrophénol, avec une structure chimique assez similaire à l'acétanilide et disponible à bas prix. Carl Duisberg, responsable de la recherche et des brevets chez Bayer AG (Friedrich Bayer & Co), demanda à son équipe de trouver une exploitation intéressante pour le paranitrophénol. Oscar Hinsberg eut l'idée de le transformer[alpha 2] en acétophénitidine[16]. La démarche de création de cette substance fut purement commerciale et par chance des tests montrent qu'elle semble plus puissante que l'antifébrine et provoque moins d'effets indésirables. Duisberg décide de mettre la nouvelle molécule en production et de la commercialiser à partir de 1888[22] sous le nom de marque « phénacétine »[23].
L'acétanilide et la phénacétine, n'ayant pas la même rapidité ni la même durée d'action, permettaient aux praticiens d'ajuster leurs prescriptions[24].
Cependant, l'acétanilide est très toxique et de nombreuses recherches se consacrent à l'élaboration de dérivés mieux tolérés. Le paracétamol fut trouvé dans les urines des personnes ayant consommé de la phénacétine. En 1889, le scientifique allemand Karl Morner découvre qu'un fragment de la phénacétine, l'acétaminophène, est un produit efficace contre la douleur et la fièvre. Une étude métabolique de ce médicament montre qu'il s'agit d'un métabolite déséthylé de la phénacétine. Cette hypothèse fut formulée dès 1894, mais fut largement ignorée à l'époque[25]. En 1893, un médecin allemand, Joseph von Mering, compare les propriétés antalgiques et antipyrétiques du paracétamol et de la phénacétine ainsi que leurs toxicités respectives. Il tire de cette étude la conclusion, erronée, que le paracétamol est plus néphrotoxique que la phénacétine : la notoriété de Von Mering fera que ce jugement ne sera pas contesté, de sorte que le paracétamol sera délaissé pendant un demi-siècle. La phénacétine sera largement employée dans les névralgies sous le nom de Veganine[26]. La toxicité de la phénacétine pour le rein sera démontrée par la suite, entraînant son retrait du marché.
L'acétanilide et la phénacétine sont en concurrence avec l'aspirine jusqu'à la fin de la Seconde Guerre mondiale. En 1938, la Food and Drug Administration, qui applique des règles plus strictes à la suite de l'adoption, cette même année, du Federal Food, Drug, and Cosmetic Act, retire brièvement du marché la phénacétine qui a été suspectée d'être cause d'agranulocytose[27].
Les travaux de David Lester et Léon Greenberg de l'université Yale[28] et ceux de Flinn et Brodie de l'université de New York confirment l'hypothèse de Karl Morner[25] - [29].
En 1946, l'Institute for the Study of Analgesic and Sedative Drugs propose une bourse au New York City Department of Health afin d'étudier les problèmes associés aux agents analgésiques. Bernard Brodie et Julius Axelrod sont désignés pour étudier le lien présumé entre les agents non dérivés de l'aspirine et le développement de la méthémoglobinémie. En 1948, ils publient leur étude[30] qui démontre que l'acétanilide est dégradé dans l'organisme en N-acétyl p-aminophénol, et que seul ce métabolite est actif contre la douleur[31].

Ils démontrent également que l'administration d'acétanilide est responsable de la formation de méthémoglobine, mais ils ajoutent que l'agent responsable est peut-être la phénylhydroxylamine, et non pas le paracétamol comme on le croyait auparavant[32]. Ils suggèrent donc aux industriels de remplacer l'acétanilide, responsable de la méthémoglobinémie, par l'acétaminophène[33]. Il y a alors un regain d'intérêt pour le paracétamol, du fait de ses propriétés antalgiques et antipyrétiques, et de sa bonne tolérance apparente.
En , ces résultats et d'autres furent présentés à New York lors d'un symposium organisé par le Institute for the Study of Analgesic and Sedative Drugs. Peu de temps avant, au vu des études des équipes de Yale et de New York, des sociétés pharmaceutiques américaines avaient commencé à produire quelques spécialités à base de paracétamol sans chercher toutefois à en pousser la vente, car elles vendaient déjà de l'aspirine. En 1950, le Triagesic est commercialisé aux États-Unis : ce mélange de paracétamol, d'aspirine et de caféine fut cependant la cause de trois cas graves d'agranulocytose ; le Triagesic fut retiré du marché en 1951 avant que l'on ne s'aperçoive que le paracétamol n'y était pour rien[34]. En 1953, les laboratoires Sterling-Winthrop Co sont les premiers à commercialiser le paracétamol sous l'appellation Panadol sur le marché britannique[35] ; producteurs d'aspirine, ils ne cherchent pas à introduire le Panadol aux États-Unis. Ce sont les laboratoires MacNeil qui vont saisir l'importance des découvertes et déposer une demande d'autorisation de mise sur le marché qui leur sera accordée par la Food and Drug Administration en 1955[alpha 3]. En 1955[36], les McNeil Laboratories, une entreprise de Pennsylvanie, lancent le Tylenol Children's Elixir : c'est un sirop pour enfant contre la fièvre et la douleur, présenté dans une boîte rouge en forme de camion de pompier[37] et qui était disponible sans ordonnance à partir de 1960[34] - [38].
Le symposium de New York avait souligné un effet secondaire de l'aspirine jusqu'alors non remarqué – que ne provoque pas le paracétamol : l'irritation de l'estomac. C'est cet avantage comparatif en faveur du paracétamol qui avait décidé les laboratoires MacNeil (qui ne produisaient pas d'aspirine) à lancer ce nouveau médicament dont la promotion sera précisément axée sur cette particularité[39]. Le produit devient ensuite populaire chez les adultes pour la même raison. En 1956, au Royaume-Uni, le paracétamol est vendu — seulement sous ordonnance — sous le nom de Panadol en dose de 500 mg, produites par Frederick Stearns & Co, une filiale de Sterling Drug Inc. En 1958 apparaît Panadol Elixir, une version destinée à l'usage des enfants[40]. Le suffixe -dol à la fin du nom du médicament provient du latin dolor, qui signifie « douleur ». Le Panadol intègre la Pharmacopée britannique en 1963[34]. En France, le paracétamol, associé à un antihistaminique, apparaît dans la spécialité Algotropyl, réservée à l'usage pédiatrique en 1957, commercialisée par les Laboratoires Bottu. Puis, la même firme pharmaceutique met sur le marché le Doliprane dès 1964[41]. Le succès de ce produit, aujourd'hui propriété de Sanofi, ne sera pas immédiat dans les pharmacies françaises : il viendra après le choix d'un mode de distribution en direct (Cooper) et par le lancement de la gamme pédiatrique en 1981. De nos jours, de nombreux médicaments contenant du paracétamol ont été développés et commercialisés dans beaucoup de pays.
En 1984, une prodrogue injectable de paracétamol est mise au point, offrant un traitement analgésique en postopératoire pour les patients ne pouvant recourir à la voie orale. Ce Pro-Dafalgan doit cependant être préparé au lit du malade ; le Perfalgan, qui ne présente pas cet inconvénient, sert à faciliter l'emploi de cette molécule pour cet usage. Associé à la morphine, il permet d'en diminuer notablement la consommation[42] (une récente étude vient toutefois relativiser l'utilité de cet emploi en association[43]).
Chimie
Structure et réactivité
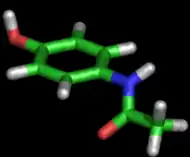
Le paracétamol est entièrement synthétique, sa formule brute est C8H9NO2. Dans les conditions ordinaires, le paracétamol est une poudre blanche avec un léger goût, soluble[44] dans 70 volumes d'eau, 7 volumes d'alcool à 95 %, 13 volumes d'acétone, 40 volumes de glycérol ou 50 volumes de chloroforme. Cependant, il est insoluble[44] dans l'éther et le benzène. Le paracétamol est stable dans l'eau, mais sa stabilité diminue en milieu acide ou basique. Les mélanges de paracétamol sont stables dans des conditions humides. Cependant, les comprimés qui contiennent de la codéine ou du stéarate de magnésium se dégradent en diacétyl-p-aminophénol dans une atmosphère humide[44].
La molécule est constituée d'un cycle benzénique, substitué par un groupe hydroxyle et par un groupe amide en position para. Le paracétamol ne comporte pas de centre stéréogène et n'a pas de stéréoisomère. Un des deux doublets libres de l'atome d'oxygène du groupe hydroxyle, le cycle benzénique, le doublet libre de l'atome d'azote et l'orbitale p du carbone du carbonyle forment un système conjugué. Cette conjugaison réduit la basicité des oxygènes et de l'azote et rend le groupe hydroxyle plus acide (comme les phénols) car la délocalisation des charges s'effectue sur un ion phénolate.
La présence de deux groupes activants rend le cycle hautement réactif pour une substitution électrophile aromatique, les substituants étant ortho et para directeurs. Toutes les positions du cycle sont plus ou moins activées de la même manière et il n'y a donc pas de site privilégié dans le cas d'une substitution électrophile. Le paracétamol est le métabolite actif de l'acétanilide et de la phénacétine : le paracétamol est produit par la décomposition de ces deux produits dans l'organisme. Ces espèces chimiques sont de la même famille chimique et ont une structure chimique très proche.
Synthèse
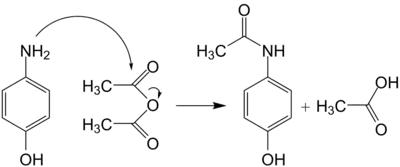
Le paracétamol ne comprend pas de centre stéréogène et n'a aucun stéréoisomère. La synthèse n'a pas besoin d'être stéréocontrôlée et elle est plus simple que les synthèses asymétriques d'autres substances pharmaceutiques.
Le paracétamol fut synthétisé pour la première fois en 1878 par Harmon Northrop Morse. La première étape est la réduction du paranitrophénol en para-aminophénol en présence d'étain dans de l'acide acétique glacial[45]. Le para-aminophénol obtenu est ensuite acylé par l'acide acétique pour obtenir du paracétamol. Vignolo simplifia cette synthèse en utilisant le para-aminophénol comme produit de départ[45]. Une seule étape d'acylation est nécessaire pour obtenir le produit désiré, ce qui raccourcit la synthèse. Plus tard, Friedlander modifia la synthèse en faisant l'acylation du para-aminophénol à partir de paranitrophénol avec de l'anhydride acétique[45] au lieu de l'acide acétique, ce qui donne un meilleur rendement.
Équation de la synthèse : C4H6O3 + C6H7NO → C8H9NO2 + CH3COOH.
L'intérêt du paracétamol a été réduit lors des premières années de commercialisation en raison d'une contamination par le para-aminophénol à cause du procédé de fabrication[25]. Cette impureté était, comme l'acétanilide, méthémoglobinisante[25].
De nos jours, il existe différentes méthodes de synthèse industrielle[45], la plupart utilisant l'acylation du para-aminophénol avec de l'anhydride acétique.
Autres dénominations
- Acetaminophen (nom utilisé aux États-Unis, Amérique latine, Japon, Canada, Corée du Sud, Iran et Hong-Kong)
- Acétyl paraminophénol, acétyl-p-amino-phénol, hydroxy-4' acétanilide, para-acétamidophénol, para-acétamino-phénol, N-acétyl-para-aminophénol.
Mécanisme d'action et devenir dans l'organisme
Mécanisme d'action
Le mécanisme d'action complet du paracétamol reste inconnu, un siècle après sa découverte[46]. Cependant, il a été démontré qu'il agit principalement au niveau du système nerveux central[47]. Selon une étude de 2006, le paracétamol agirait en inhibant au niveau central la production de prostaglandines, impliquées dans les processus de la douleur et de la fièvre, par le biais d'une action inhibitrice sur l'enzyme prostaglandine H2 synthase (PGHS), qui comporte notamment un site actif « cyclooxygénase » (ou COX), cible de la majorité des anti-inflammatoire non stéroïdien (AINS), et un site « peroxydase » (ou POX), sur lequel agirait le paracétamol[48]. Le paracétamol n'aurait pas d'action directe sur le COX-1 et le COX-2[49], les deux formes de cyclooxygénase (COX) sur lesquelles agissent les anti-inflammatoire non stéroïdien (AINS) comme l'aspirine ou l'ibuprofène. On soupçonne l'existence d'une nouvelle isoenzyme, le COX-3 (enzyme produite par épissage successif de la COX-1 entraînant un décalage de lecture des bases par le corps humain), sur laquelle agirait spécifiquement le paracétamol[50] et qui expliquerait pourquoi le paracétamol réduit la fièvre et la douleur tout en étant dénué d'activité anti-inflammatoire et antiplaquettaire. Pour l'instant, cette hypothèse n'a pas été prouvée chez l'humain[47]. D'autres mécanismes d'action ont été évoqués pour expliquer l'activité analgésique et antipyrétique du paracétamol. Un mécanisme d'action sérotoninergique central est suspecté depuis quelque temps[51]. Le paracétamol potentialiserait l'effet des neurones sérotoninergiques descendants de la moelle épinière exerçant un contrôle inhibiteur sur les voies de la douleur. Par ailleurs, le paracétamol pourrait agir en limitant la libération de Béta-endorphines[52].
Les recherches récentes montrent que l'ion peroxynitrite pourrait être la source oxydante permettant aux COX de transformer l'acide arachidonique en prostaglandine[53] - [54]. De même que la nitrotyrosine (en)[55] est un marqueur spécifique de l'excès de peroxynitrites agissant comme agent nitrant sur le cycle phénolique activé de la tyrosine, le nitroparacétamol[56] formé par nitration directe du paracétamol par les peroxynitrites les consommerait et permettrait d'annihiler la synthèse des prostaglandines. Ces hypothèses ont été confirmées par Schildknecht et al.[57]
Pharmacocinétique
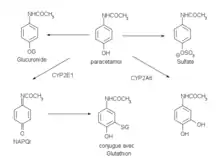
L'absorption du paracétamol par voie orale est complète et rapide : le maximum de concentration plasmatique est atteint entre 15 minutes (comprimé effervescent) et 30–60 minutes (comprimé et poudre) après ingestion.
Le paracétamol se distribue rapidement dans tous les tissus. Les concentrations sont comparables dans le sang, la salive et le plasma. Le paracétamol est métabolisé essentiellement au niveau du foie. Les deux voies métaboliques majeures sont la glycuroconjugaison et la sulfoconjugaison. Il existe une voie métabolique moins importante catalysée par le Cytochrome p450 (plus précisément par les isoenzymes CYP2E1, CYP1A2, CYP3A4)[58], qui aboutit à la formation d'un intermédiaire réactif toxique, la N-acétyl-p-benzoquinone imine ou NAPQI. Il est normalement rapidement éliminé par réaction avec le glutathion réduit puis évacué dans les urines après conjugaison à la cystéine et à l'acide mercaptopurique.
L'élimination du paracétamol est essentiellement urinaire : 90 % de la dose ingérée est éliminée par le rein en 24 heures, principalement sous forme glycuroconjuguée (60 à 80 %) et sulfoconjuguée (20 à 30 %) et moins de 5 % est éliminé sous forme de paracétamol. La demi-vie d'élimination est d'environ deux heures.
Variations physiopathologiques
En cas d'insuffisance rénale sévère, avec une clairance de la créatinine inférieure à 10 mL/min, l'élimination du paracétamol et de ses métabolites est retardée. La glycuroconjugaison est immature chez le nourrisson et l'enfant, le paracétamol est donc essentiellement sulfoconjugué. Le passage à une voie métabolique identique à celle de l'adulte intervient entre 9 et 12 ans[59].
Galéniques, association et dénominations commerciales

Formes galéniques
Le paracétamol entre dans la composition d'une soixantaine de spécialités pharmaceutiques et peut se présenter sous différentes formes ou conditionnements. Le paracétamol seul est vendu sous de nombreuses formes galéniques[60] comme des comprimés, en poudre, des comprimés effervescents, des comprimés orodispersibles, des gélules, du sirop, des suspensions buvables, des suppositoires pour adultes ou enfants, ou des lyophilisats. Il est aussi disponible sous forme intraveineuse.
Association
Le paracétamol peut être associé à d'autres antalgiques au sein d'un même médicament, dans le but principal d'améliorer l'efficacité globale et d'optimiser le rapport bénéfice/risque en diminuant les posologies, mais aussi afin d'allonger la durée d'action, d'élargir le spectre d'efficacité, de diminuer l'accoutumance, d'améliorer l'observance et de minimiser le risque d'usage détourné. Le but des associations de médicaments étant de produire des interactions pharmaceutiques bénéfiques, c'est-à-dire une synergie, permettant d'augmenter l'efficacité et d'améliorer la tolérance tout en utilisant les doses les plus faibles possibles. L'association doit permettre d'élargir le spectre d'efficacité en combinant des antalgiques agissant simultanément sur des cibles différentes, mais impliqués dans des mécanismes physiopathologiques identiques[61].
Le paracétamol est utilisé en association avec d'autres substances actives pour profiter de ses propriétés antalgiques et antipyrétiques. L'un des problèmes des associations est l'accumulation des effets secondaires ; cependant, le paracétamol étant très bien toléré, il est particulièrement intéressant dans le cadre des associations, et c'est pourquoi les laboratoires pharmaceutiques ont développé de très nombreuses formules comprenant du paracétamol. Un dérivé lipidique, le palmitostéarate de glycérol atomisé est parfois ajouté aux mélanges pour masquer le goût du paracétamol[62].
Dénominations commerciales
Le paracétamol non associé est vendu en nom générique ou sous de nombreuses marques dont certaines très connues :
- Doliprane (Sanofi, médicament le plus prescrit en France), Dafalgan (UPSA, deuxième médicament le plus prescrit en France), Efferalgan (UPSA, troisième médicament le plus prescrit en France) et de nombreuses autres marques en France. En vente libre (c'est-à-dire sans ordonnance, prescrit directement par le pharmacien), le prix d'un traitement d'une semaine est généralement réglementé à 2,08 € (100 mL de sirop enfant) ou 1,94 € (adulte, huit fois 1 000 mg ou seize fois 500 mg) (France, 1,12 € prix public TTC plus 0,82 € de rémunération forfaitaire du pharmacien. Le pharmacien et les employés polyvalents de pharmacie ne sont pas rémunérés à l'acte de conseil mais à la quantité de médicaments prescrits par le médecin ou par le pharmacien).
- Dafalgan, Mylan, Perdolan, Sandoz et de nombreuses autres marques en Belgique. En vente libre (c'est-à-dire sans ordonnance, prescrit directement par le pharmacien), le prix d'un traitement est entièrement libre, il est généralement constaté à 7,45 € (200 mL de sirop enfant, soit 79 % plus cher qu'en France) ou 3,24 € (adulte, le prix varie du simple au triple selon les marques et les conditionnements, le patient devant faire des demandes écrites pour les conditionnements les moins chers s'il souffre d'une maladie chronique), dix fois 1 000 mg ou vingt fois 500 mg, soit 34 % plus cher qu'en France)[63].
- Tylenol ou Panadol au Canada et aux États-Unis, le prix est libre et un exemple sur internet est à 8,99 USD (7,47 €) (adulte, cent fois 325 mg, soit 7 % moins cher qu'en France).
- Alors que les médicaments sont généralement plus chers en Allemagne qu'en France, le générique allemand Ratiopharm est à 1,57 €, prix libre moyen constaté (adulte seize fois 500 mg, soit 19 % moins cher qu'en France). La même boîte est théoriquement vendue 1,90 € en France (soit 2 % moins cher que le tarif réglementé), mais lorsque le patient demande un paracétamol générique en France, il obtient le plus souvent de l'UPSA (1,94 €)[64].
On le retrouve associé à d'autres substances actives dans certains remèdes contre les états grippaux (Actifed, Dolirhume, Fervex, Humex Rhume, Rhinofébral), où il est efficace à la fois sur la fièvre et la douleur. Il est parfois mélangé avec de la caféine (Dalféine, Prontadol, Claradol caféiné, Exidol, Theinol), substance qui pourrait augmenter son effet analgésique, mais cette notion reste très controversée[65] - [66] - [67]. Il peut également être associé à d'autres antalgiques tels que l'aspirine (Novacétol) et on le retrouve souvent associé à un opiacé comme la codéine (Dafalgan Codéine, Codoliprane), le dextropropoxyphène (spécialités retirées), la poudre d'opium (Lamaline), le tramadol (Ixprim, Zaldiar), ce qui permet d'augmenter son action antalgique et de traiter les douleurs moyennes ou fortes[68]. Depuis 2011, le dextropropoxyphène, seul ou en association, a été retiré du marché français. Il existait de nombreuses présentations de l'association paracétamol-dextropropoxyphène (Dialgirex, Di-antalvic). L'efficacité clinique antalgique (en termes de synergie de l'analgésie) de l'association paracétamol + dextropropoxyphène reste mal évaluée (à la différence de celle utilisant la codéine). Il n'est pas démontré que l'association paracétamol + dextropropoxyphène est supérieure au paracétamol seul[69].
Les présentations de l'association paracétamol-tramadol comportent 37,5 mg de tramadol et 325 mg de paracétamol par comprimé, ce qui permettrait d'obtenir une efficacité antalgique équivalente à 50 mg de tramadol mais avec une meilleure tolérance. L'association du paracétamol avec un opiacé peut poser des problèmes de dépendance et de détournement d'usage.
Indications, posologie et informations pratiques

Indications
Le paracétamol est utilisé pour[70] :
- le traitement symptomatique des douleurs aiguës ou chroniques, d'intensité légère à modérée[71]. Il s'agit d'un antalgique de palier 1 selon la classification de l'OMS[72]. Il peut être utilisé seul ou en association avec d'autres antalgiques (codéine, tramadol, acide acétylsalicylique, ibuprofène), il rentre alors dans la classification des antalgiques de palier 2 indiqués dans les douleurs d'intensité modérée à intense ou ne répondant pas à l'utilisation d'antalgiques périphériques seuls ;
- le traitement symptomatique de la fièvre, en particulier chez l'enfant chez qui il constitue l'antipyrétique de première intention[71] - [73]. La revue EvidenceBasedMedecine s'est toutefois interrogée sur la solidité des données attestant de son efficacité comme antipyrétique[74] tandis que par ailleurs l'opportunité du traitement systématique de la fièvre a pu être questionnée dans un bulletin de l'Organisation mondiale de la santé (OMS) de 2003[75]. Il y était affirmé : « Aucune étude ne montre d'avantage manifeste du paracétamol à dose thérapeutique chez l'enfant fébrile atteint d'infection virale ou bactérienne ou de paludisme. D'après certaines études, la fièvre semblerait même avoir un effet bénéfique sur l'infection, bien qu'aucune étude prospective définitive n'ait été réalisée chez l'enfant pour tester cette hypothèse. Ce traitement ne devrait donc être administré qu'à l'enfant manifestement incommodé ou dont l'affection est douloureuse[76]. Le , l'American Academy of Pediatrics, a réitéré ces recommandations[77]. »
Posologie
La dose, ou posologie, maximale peut varier d'un pays à l'autre selon la recommandation des produits de santé. En France, la recommandation est de[78] - [70] :
- adultes : 500 à 1 000 mg par prise, en espaçant les prises de quatre heures minimum. Il n'est généralement pas nécessaire de dépasser la dose de 3 g/j mais exceptionnellement (en cas de douleurs intenses non complètement contrôlées par 3 g/j, et sur avis médical), on peut atteindre un maximum de 4 g/j (soit quatre fois 1 000 mg ou huit fois 500 mg) ;
- enfants : la dose quotidienne recommandée est de 60 mg/kg par jour, à répartir en quatre ou six prises, soit environ 15 mg/kg toutes les six heures ou 10 mg/kg toutes les quatre heures. La dose maximale est de 80 mg/kg par jour chez l'enfant de moins de 38 kg selon les recommandations officielles en France.
Informations pratiques
- En France, en Belgique, en Suisse, au Royaume-Uni, aux États-Unis et au Canada, le paracétamol peut être délivré en pharmacie sans ordonnance ou sur prescription médicale.
- En cas d'oubli de la dernière prise, on peut reprendre le médicament aussitôt puis continuer selon la posologie prescrite, mais en respectant un intervalle de quatre heures entre chaque prise.
- Les gélules et les comprimés sont à avaler tels quels avec une boisson comme de l'eau, du lait ou un jus de fruit.
- Pour les comprimés effervescents, laisser dissoudre complètement le comprimé dans un verre d'eau, si besoin après l'avoir cassé en deux parts égales s'il est sécable.
Depuis 2011, l'association paracétamol + dextropropoxyphène (Di-Antalvic) est retirée du marché[79]. Ce retrait est surtout lié à des surdosages, lors d'intoxications volontaires (tentatives de suicide) et non à une toxicité à dose thérapeutique[80].
Contre-indications, précautions d'emploi et effets indésirables
Contre-indications
Les contre-indications absolues sont[70] l'hypersensibilité au paracétamol, l'insuffisance hépato-cellulaire sévère et la porphyrie.
On peut retrouver de l'aspartame dans certaines formes commerciales ; dans ce cas le médicament est contre-indiqué en cas de phénylcétonurie.
Précautions d'emploi
Le paracétamol est autorisé en cas de grossesse et d'allaitement. Cependant plusieurs études ont mis en cause cette molécule. En 2016, une étude publiée dans JAMA Pediatrics (en) a mis en évidence un lien entre prise de paracétamol pendant les second et troisième trimestres de la grossesse et l'augmentation du risque de certains troubles du comportement comme l'hyperactivité[81]. Les auteurs restent toutefois très prudents et n'écartent pas l'hypothèse qu'un autre facteur, lié à la prise de paracétamol, pourrait expliquer l'effet neurologique observé.
Il pourrait aussi exister une relation entre la prise de paracétamol pendant la grossesse et plus spécialement au cours du premier trimestre, et le risque pour les enfants de souffrir de problèmes respiratoires ou d'asthme avant l'âge de 7 ans[82]. L'effet serait toutefois faible, avec une augmentation de risque de 11 à 22 % et ne concerne que la prise durant la grossesse ou la petite enfance[83]. Comme dans le cas des troubles du comportement les scientifiques ne peuvent exclure que cette augmentation du risque d'asthme ne soit pas liée aux infections motivant la prise de paracétamol.
Perturbateur endocrinien de par un effet anti-androgène[84], la prise de paracétamol, d'autant plus sur de longues périodes (une à quatre semaine selon différentes études[85]) et en association avec d'autres analgésiques, pourrait constituer durant la grossesse un facteur de risque d'anomalies du développement de l'appareil reproducteur masculin (cryptorchidisme)[86] - [87].
Pendant la période d'allaitement, le paracétamol passe dans le lait maternel. Toutefois, les quantités excrétées dans la lactation sont inférieures à 2 % de la quantité ingérée et le paracétamol n'est donc pas contre-indiqué pendant la période d'allaitement[70]
À part avec certains anticoagulants oraux et les sétrons (antiémétiques), il n'y a aucune interaction médicamenteuse particulière répertoriée pour le paracétamol.
- Anticoagulant oral : le paracétamol, utilisé à des doses supérieures à 3 g/j, pendant plus de 4 jours consécutifs, pourrait potentialiser l'activité anticoagulante des Anti-Vitamine K (AVK)[70]. Dans ce cas, une surveillance de l'INR serait recommandée[88] - [89].
- Sétrons : une compétition existe entre le paracétamol et l'odansétron notamment, ayant pour effet de diminuer l'efficacité antalgique du paracétamol[90].
La prise de paracétamol peut fausser[70] le dosage de l'acide urique sanguin par la méthode à l'acide phosphotungstique, ainsi que le dosage de la glycémie par la méthode à la glucose oxydase-peroxydase.
Il n'y a aucune interaction alimentaire rapportée pour le paracétamol[70].
Pour éviter tout risque de surdosage, il faut vérifier l'absence de paracétamol dans la composition d'autres médicaments pris de façon concomitante.
Effets indésirables
Habituellement le paracétamol est très bien toléré lorsqu'il est pris à des doses thérapeutiques[91]. Des effets indésirables ont néanmoins été rapportés sans que l'imputabilité (le fait que l'effet indésirable soit bien causé par le médicament) ait été établie la plupart du temps. Les principaux effets indésirables retrouvés dans la littérature sont :
- Très rarement : éruption cutanée avec rash ou éruption urticarienne d'origine probablement allergique[92] - [93], thrombopénie[94] et asthme[95] - [96].
- Controversé : hépatite aiguë cytolytique[97] - [98] - [alpha 4] et insuffisance rénale chronique[91] - [alpha 5].
- De façon ponctuelle : hypotension[99] - [alpha 6], choc anaphylactique[100] - [101], purpura vasculaire[102], syndrome de Lyell et syndrome de Stevens-Johnson[103], ulcération rectale[104], agranulocytose[105], pancréatite aiguë généralement en association avec d'autres médicaments comme la codéine[106] - [107], hépatite chronique active[108], hépatite granulomateuse[109] et rhabdomyolyse[110].
Une toxicité sur le foie à dose thérapeutique ne peut également être exclue chez certaines personnes à risques[111] - [112].
Chez le très jeune enfant, l'administration de paracétamol pourrait augmenter le risque de survenue d'un asthme[113] - [114].
A la dose de 4 grammes par jour, le paracétamol semble augmenter légèrement la pression artérielle, sans preuve d'une conséquence médicale[115].
In vitro, le paracétamol pourrait présenter un effet tératogène[116] qui n'est pas présent in vivo. La molécule n'est donc pas contre-indiquée chez la femme enceinte.
Devant l'apparition d'un effet indésirable, il est nécessaire d'arrêter le médicament incriminé et de consulter son médecin.
Surdosage
Le paracétamol est un médicament utilisé couramment et disponible dans les pharmacies. Les cas de surdose sont courants — plus de 100 000 par an aux États-Unis, une centaine en Suisse[117] — et ont des conséquences graves. Ils peuvent entraîner, si le surdosage est au long terme (environ 10 g/jour pendant deux semaines), une nécrose hépatique ; dans le cas d'un surdosage rapide (25–30 g en un jour), une hépatite fulminante, qui nécessite une transplantation immédiate pour éviter le décès du patient. En France, la mention « surdosage = danger - dépasser la dose peut détruire le foie » est obligatoire sur les boîtes de paracétamol depuis 2020[118].
Dose toxique
La dose toxique du paracétamol est hautement variable selon les individus. En une prise unique, elle est de l'ordre de 10 g ou 150 mg/kg chez l'adulte et de 150 mg/kg chez l'enfant[119] - [120]. Certains ne parlent d'intoxication aiguë que pour des doses élevées supérieures à 200 mg/kg, soit plus de 14 g pour un humain de 70 kg[121].
Cependant, le paracétamol peut être toxique pour le foie même à 4 g/24 h, soit des doses thérapeutiques[122]. Administrées sur de longues périodes, ces doses se rapprochent des doses toxiques pouvant entraîner des lésions hépatiques permanentes voire mortelles, surtout chez des patients à la fonction hépatique préalablement altérée[121] - [123] - [124]. C'est notamment le cas de personnes souffrant de maladies hépatiques ou d'alcoolisme chronique provoquant une diminution des réserves de glutathion[125] - [126]. Par contre, seuls quelques rapports font état d'une toxicité du paracétamol lors de situations diminuant les réserves de glutathion, comme une infection par le VIH, une hépatite chronique C ou une cirrhose hépatique par exemple[127].
Christophe Bureau, secrétaire général de l'Association française pour l'étude du foie (Afef) explique que pour les personnes à risque, « dépasser de façon très légère les doses journalières peut être mortel… C'est la première cause d'hépatite fulminante dans le monde, c'est-à-dire de destruction massive du foie, qui nécessite une greffe d'urgence, il a tué 400 personnes aux États-Unis en 2009. Je ne comprends pas qu'il soit encore en vente libre »[128].
Ainsi sur de longues durées, la différence entre une dose thérapeutique et une dose toxique serait faible. Depuis que le paracétamol est présent dans de nombreux médicaments, mélangé à d'autres molécules, le risque de surdosage involontaire est majoré[121]. Les prises de paracétamol doivent toujours être espacées de quatre heures au minimum. Pour éviter le surdosage, il est utile de discuter avec un pharmacien pour connaître les médicaments contenant du paracétamol ou bien de regarder la composition des médicaments pour détecter la présence de paracétamol.
Risques et tableau clinique
Une des étapes de la métabolisation du paracétamol produit une molécule toxique, la N-acétyl-p-benzoquinone imine (ou NAPQI), via les cytochromes P450 (CYP2E1, CYP1A2, CYP3A4). Ce métabolite peut provoquer la mort des cellules hépatiques. Il est éliminé, dans le foie, par une réaction avec le glutathion (donneur de SH) qui capte les radicaux. Aux doses thérapeutiques recommandées, la NAPQI est éliminée par l'organisme et ne représente pas un danger. Par contre, lorsque la dose de paracétamol est trop importante, la NAPQI est produite en grande quantité, les réserves de glutathion s'épuisent et le foie n'arrive plus à l'éliminer ; il subira des dommages plus ou moins importants selon la quantité de paracétamol absorbée. Un risque accru de toxicité est provoqué par un manque de glutathion (malnutrition, anorexie, éventuellement maladies du foie) ou une formation accrue du métabolite toxique.
Le surdosage en paracétamol peut ainsi entraîner une hépatite avec de graves lésions du foie (cytolyse hépatique), conduisant à une nécrose dans les cas extrêmes. Les conséquences d'un surdosage sont graves, parfois mortelles. Les dommages causés au foie sont irréversibles, une greffe de foie devenant nécessaire lorsque les dommages sont très importants. La NAPQI entraîne la création d'adduits fixés aux protéines hépatiques, dégradation des lipides membranaires, perturbations de l'homéostasie calcique, provoquant une nécrose et une hépatite cytolytique. Le rein est touché par le même mécanisme.
La toxicité sur le foie est prédictible à l'aide de deux paramètres : la dose ingérée et le taux plasmatique du paracétamol (ou paracétamolémie). Les prises intentionnellement abusives de paracétamol peuvent être détectées rapidement et les dommages peuvent être limités par l'administration de N-acétylcystéine. Ce n'est pas le cas de surdosages non intentionnels et chroniques qui se détectent plus tardivement alors que des dommages importants ont déjà pu se produire.
De plus, il est possible de calculer la demi-vie d'élimination du paracétamol. Dans les cas d'intoxication, la nécrose hépatique empêche l'élimination et la demi-vie augmente. Une demi-vie supérieure à quatre heures témoigne d'une hépatite. Une demi-vie supérieure à douze heures indique une insuffisance hépato-cellulaire.
Les individus qui ont pris trop de paracétamol n'ont généralement pas de symptômes pendant les vingt-quatre premières heures[129]. Bien que des nausées ou des vomissements apparaissent en premier, ces symptômes disparaissent après quelques heures. Les sujets se sentent mieux et croient que le pire est passé. Si la dose absorbée est toxique, après cette période de bien-être, le sujet a une défaillance hépatique. Dans les cas extrêmes, le sujet tombe dans le coma avant d'avoir une défaillance du foie[129].
Les enfants supportent mieux le paracétamol, car ils possèdent un foie et des reins plus larges par rapport à la taille de leur corps, et ils sont plus tolérants à ce produit[130]. La demi-vie sera plus importante chez l'enfant qui possède des capacités de glucuronoconjugaison inférieures à celles de l'adulte[60]. Les preuves à l'heure actuelle sont insuffisantes pour conclure que l'utilisation régulière de paracétamol est associée à un risque accru d'insuffisance rénale chronique[49].
Une surdose massive de paracétamol, habituellement plus de 40 g, peut également entraîner une acidose lactique métabolique ; celle-ci s'installe avant la cytolyse hépatique[131]. Le paracétamol et certains de ses métabolites inhibent la respiration cellulaire, conduisant à l'accumulation de lactates.
Prise en charge
Toute personne ayant ingéré une dose supérieure à la dose toxique théorique ou ayant ingéré une dose inconnue supposée supérieure, doit être immédiatement transférée dans un service d'urgences hospitalier[132] où le traitement peut être l'administration en intraveineuse ou orale de N-acétylcystéine.
L'absorption du paracétamol par voie gastro-intestinale est complète au bout de deux heures en conditions normales, donc une décontamination gastro-intestinale n'est utile que pendant ce laps de temps. L'absorption du paracétamol peut être retardée en cas d'ingestion de nourriture. L'absorption est plus rapide lorsque le paracétamol est sous forme soluble que sous la forme solide.
Le lavage gastrique n'est pas recommandé[70], tout comme les vomissements provoqués, par l'utilisation d'un vomitif[133]. Le sirop d'ipéca doit notamment être considéré comme obsolète[134].
Le charbon activé, qui réduit l'absorption digestive du paracétamol et présente moins de risques que le lavage gastrique, est indiqué uniquement lorsque la quantité de paracétamol absorbée est potentiellement mortelle et que l'ingestion a eu lieu moins d'une heure avant[135]. Dans ce cas, on recommande une dose unique de 1 à 2 g/kg d'une suspension aqueuse de charbon actif administrée par voie orale[58]. Auparavant, les médecins étaient réticents à administrer du charbon activé puisqu'en cas de surdosage, celui-ci peut absorber aussi l'antidote et donc diminuer son efficacité. Mais des études ont montré que l'adsorption d'une partie de la N-acétylcystéine orale par le charbon activé n'a pas de conséquences significatives[70], l'une d'entre elles a déterminé que seulement 39 % de la N-acétylcystéine est absorbée lorsqu'elle est administrée en même temps que le charbon[136]. Sinon, l'utilisation d'acétylcystéine par voie intraveineuse est efficace en combinaison avec du charbon activé. S'il est prévu de donner la N-acétylcystéine par voie orale, il est recommandé de différer le traitement de une à deux heures après l'administration de charbon activé[58].
En pratique clinique, la prise en charge est la suivante : recherche d'intoxications associées[137], prise des signes vitaux (pouls, pression artérielle, température, score de Glasgow, fréquence respiratoire) et prélèvement veineux (paracétamolémie, transaminases, taux de prothrombine, créatinine, ionogramme)[137], pose d'une voie veineuse périphérique avec une solution polyionique type B26 : 2 l/24 h[137], charbon activé si la prise est inférieure à deux heures[137]. Puis l'administration de N-acétylcystéine dépend de la dose ingérée :
- en cas de dose ingérée connue inférieure à la dose toxique minimale, il n'y a pas de traitement nécessaire car pas d'intoxication sérieuse[58] ;
- si la dose supposée ingérée est inférieure à 8 grammes, l'administration est guidée par la paracétamolémie et le délai écoulé depuis la prise de paracétamol. Le choix du traitement est déterminé selon les abaques de Prescott (ou nomogramme de Rumack-Matthew)[138] ;
- si la dose supposée ingérée est supérieure à huit grammes, l'administration de N-acétylcystéine est immédiate, « à l'aveugle », sans attendre les résultats du taux plasmatique de paracétamol[138]. Quatre heures au minimum après l'ingestion, il faut déterminer la paracétamolémie et la rapporter aux abaques de Prescott. Si le taux se situe en dessous de la « ligne de traitement », on peut arrêter le traitement. Si le taux est au-dessus, il faut le continuer et l'appliquer entièrement. Chez les patients à risque (affection hépatique, alcoolisme chronique, induction du métabolisme hépatique, malnutrition), il convient d'appliquer le schéma complet même en cas de dose plus faible[58]. Le nomogramme ne peut pas être utilisé si le moment de l'ingestion est inconnu, s'il y a eu plusieurs ingestions ou s'il y a des facteurs de risque[58].
Le transfert en unité de réanimation est indiqué en cas de troubles hémodynamiques, neurologiques, respiratoires, de co-intoxication avec une substance exigeant une prise en charge en réanimation, d'hépatite cytolytique grave, et a fortiori, d'insuffisance hépatique[137]. En fin de traitement il faut contrôler le taux de prothrombine, les transaminases, la créatinine et la glycémie[137]. La sortie est possible si la paracétamolémie arrive dans les zones non toxiques, en l'absence de toxiques associés et après accord du psychiatre (en cas d'intoxication volontaire)[137].
N-acétylcystéine

Le métabolite responsable de la toxicité du paracétamol est la N-éthanoyl-4-hydroxyphénylhydroxylamine. Elle réagit de manière irréversible avec les thiols tels le glutathion[139] dont le foie a besoin. C'est la diminution de ce glutathion hépatique qui crée cette insuffisance hépatique. La protection apportée par la N-acétylcystéine, précurseur de la cystéine, ou diverses autres molécules comportant un groupe SH telles la méthionine ou la cystéamine, provient de ce que le métabolite toxique va réagir avec eux plutôt qu'avec le glutathion, qui sera ainsi d'épargné. Ces produits permettent de réduire le risque de toxicité sur le foie et de prévenir la nécrose hépatique par le paracétamol et s'ils sont absorbés moins de huit heures après l'ingestion du paracétamol[58].
Après huit heures, une série d'évènements toxiques dans le foie commence et le risque de nécrose hépatique et de décès augmente de façon critique. Bien que la N-acétylcystéine soit plus efficace lorsqu'elle est administrée tôt, le produit a cependant des effets bénéfiques jusqu'à 48 heures après l'ingestion[140]. Elle n'endommage pas les cellules et peut être excrétée sans danger.
La N-acétylcystéine s'administre comme antidote soit par voie buccale (Fluimucil granulé ou Mucomyst soluté, disponibles en pharmacie), soit en perfusion intraveineuse (Fluimucil 20 % (Inpharzam), amp. à 25 ml, 1 g = 5 ml). Aux États-Unis, l'administration orale est la méthode de référence alors qu'en Europe, l'administration par voie intraveineuse est préférée[58]. L'acétylcystéine par voie orale peut entraîner à cause de son goût et de son odeur soufrée, des vomissements et des nausées. Par voie intraveineuse, surtout en cas de perfusion trop rapide, elle peut entraîner des réactions anaphylactoïdes[70]. Le choix de la voie d'administration, orale ou intraveineuse, dépend avant tout de l'existence ou non de vomissements[141].
Il existe trois schémas thérapeutiques différents, un par voie orale, deux par voie veineuse, d'efficacité équivalente tant qu'ils sont instaurés dans les dix heures suivant l'ingestion[58] - [141] :
- Schéma de Prescott[142] (voie veineuse) : dose initiale de charge de 150 mg/kg (dans 200 ml de glucose 5 % sur 60 min), puis 50 mg/kg (dans 500 ml de glucose 5 % sur 4 h), puis 100 mg/kg (dans 1 000 ml de glucose 5 % sur 16 h). Dose totale de 300 mg/kg sur une durée totale de 21 h ;
- Schéma de Smilkstein[143] - [144] (voie veineuse) : dose initiale de charge de 140 mg/kg (dans 200 ml de glucose 5 % sur 15 min), puis 70 mg/kg (dans 100 ml de glucose 5 % sur 15 min) toutes les 4 h, à répéter douze fois. Dose totale de 980 mg/kg sur une durée totale de 48 h ;
- Schéma de Rumack[145] (voie orale) : dose initiale de charge de 140 mg/kg, puis 70 mg/kg toutes les 4 h, à répéter dix-sept fois. Dose totale de 1 330 mg/kg sur une durée totale de 68 h.
Si le traitement est commencé plus de dix heures après l'ingestion, le schéma d'administration orale de Rumack et le schéma d'administration intraveineuse de Smilkstein donnent de meilleurs résultats que le schéma de Prescott[58].
Comparaison avec les anti-inflammatoires non stéroïdiens et l'aspirine
Le paracétamol, contrairement à l'aspirine et à l'ibuprofène, est dépourvu de propriétés anti-inflammatoires. Il ne fait pas partie de la classe des anti-inflammatoires non stéroïdiens (AINS), n'étant pas un bon inhibiteur des COX et notamment de la COX-2. Les AINS eux, ont en commun la propriété de pouvoir diminuer la production des prostanoïdes en inhibant l'activité des deux isoformes de cyclooxygénases (COX-1 et COX-2)[146].
En ce qui concerne le traitement de la douleur, l'activité antalgique du paracétamol est comparable à celle de l'aspirine, pour des posologies identiques de 1 à 3 g/jour et pour des douleurs de causes diverses[146] - [60].
Des études renforcent la notion qu'il faut continuer à envisager le paracétamol comme traitement de première intention pour le soulagement de la douleur d'intensité légère à modérée[147] d'après des évaluations effectuées relativement à l'innocuité, à l'efficacité et au coût[147]. Le paracétamol a très peu d'effets secondaires. Les associations avec d'autres produits, plus puissantes ou mieux adaptées ne seront envisagées que dans un second temps, ou dans des cas spécifiques. Dans les doses recommandées, le paracétamol n'irrite pas la paroi de l'estomac, n'affecte pas la coagulation du sang autant que les AINS, et n'affecte pas le fonctionnement du rein. L'utilisation des AINS peut être à l'origine de cas d'hémorragies gastro-intestinales ; le paracétamol, par contre, n'est pas associé à l'augmentation du risque d'épisodes gastro-intestinaux dans les doses normales. Cependant, certaines études ont montré que pour des doses élevées (plus de 2 000 mg/j) le risque de complications intestinales augmente[148].
Le paracétamol ne présente pas de contre-indications pour les femmes enceintes et n'affecte pas le développement du fœtus comme le font les AINS (traitement de la persistance du canal artériel). L'utilisation des AINS par les femmes enceintes est associée, de façon importante, à l'hypertension pulmonaire persistante chez les nouveau-nés[149]. Le paracétamol est actuellement très utilisé, notamment en pédiatrie. Il peut être administré aux enfants car il n'est pas associé au risque du syndrome de Reye pour les enfants possédant une déficience immunitaire. Une étude clinique faite chez des enfants montre qu'une dose standard d'ibuprofène provoque un plus grand soulagement de la douleur qu'une dose standard de paracétamol ou de codéine[150]. Comme les AINS et contrairement aux opiacés, le paracétamol n'a pas été reconnu comme la cause d'euphories ou de modification d'humeur mais contrairement aux opiacés, il peut endommager le foie. Le paracétamol et les AINS présentent un faible risque d'assuétude ou d'addiction, contrairement aux opiacés.
En ce qui concerne le traitement de la fièvre, il ne semble pas exister de différence d'efficacité anti-pyrétique entre le paracétamol et les AINS[146] - [60]. Concernant l'enfant, deux méta-analyses de 2004[151] - [152] retrouvent que l'ibuprofène aurait une rapidité d'action légèrement supérieure au paracétamol. Mais c'est le paracétamol qui permettrait le mieux d'améliorer le confort de l'enfant, notamment au niveau de l'activité et de la vigilance[153]. Au total on peut conclure que chez l'enfant, le paracétamol, l'ibuprofène et l'aspirine ont une efficacité antipyrétique identique mais que leurs effets indésirables sont sensiblement différents, ce qui finalement justifie amplement de privilégier le paracétamol en première intention[154].
Paracétamol et société
Surdosage involontaire et suicide
Le surdosage involontaire en paracétamol est la première cause de défaillance du foie en Angleterre et aux États-Unis[155]. Les intoxications involontaires au paracétamol représentent tous les ans aux États-Unis plus de 13 000 passages aux urgences, plus de 2 000 hospitalisations et près de 100 décès selon la Food and Drug Administration[156]. Ces chiffres importants sont expliqués par le fait que de nombreux produits sont disponibles aux États-Unis en vente libre sans ordonnance et contiennent du paracétamol sans que cela soit indiqué sur la boîte, et par le fait que les conditionnements des antalgiques à base de paracétamol dépassent souvent la dose potentiellement mortelle de huit grammes par boîte. En France dans les années 1980, l'Agence du médicament, ancien nom de l'Afssaps, avait réduit le conditionnement des antidouleurs à base de paracétamol pour qu'ils ne dépassent pas cette dose. Depuis ce changement de conditionnement, les décès par intoxication n'ont pas augmenté alors que la consommation n'a pas cessé de croître. Ainsi en 1990, 177 420 000 boîtes de paracétamol ont été vendues en France, et 5 335 intoxications et six décès ont été recensés. Ces chiffres restent stables depuis cette année. En Angleterre, à l'époque où le conditionnement n'était pas limité à un maximum de huit grammes, les décès étaient compris entre 200 et 600 selon les sources, ce qui a mené les autorités à adopter des mesures similaires à la France à partir de 1998[157].
Le paracétamol est parfois utilisé lors de suicides ou de tentatives de suicides. Cependant, plus de la moitié des morts par surdosages sont des accidents. Les défaillances hépatiques aiguës consécutives à un surdosage non intentionnel donnent souvent des tableaux plus sévères et ont un pronostic moins bon que chez les patients ayant un surdosage intentionnel. En effet, les victimes de surdoses accidentelles sont souvent prises en charge plus tard, et les risques sont donc plus élevés. Cependant, comparés aux nombres de doses de paracétamol consommées chaque jour, les surdosages accidentels ne touchent qu'une minorité des utilisateurs. En France, les suicides au paracétamol sont bien moins courants mais aussi plus difficiles à évaluer car il n'existe pas de registre national des intoxications volontaires[158]. Bien que le taux d'intoxication au paracétamol soit faible par rapport aux millions de tablettes utilisées chaque année, certains auteurs proposent de changer le mode de vente du paracétamol. Les conditionnements actuels limitent le risque de surdosage accidentel, la quantité de paracétamol par boîte a été diminuée et les prescriptions de médicaments combinant des narcotiques au paracétamol ont été restreintes pour réduire les accidents. Les enfants sont victimes de surdoses accidentelles en cas d'absorption massive sous la forme de sirop. Par contre, les formes effervescentes du paracétamol limitent le risque de prise accidentelle, car elles imposent de boire une grande quantité de liquide. L'association d'une substance et de son antidote dans le même médicament permet de diminuer les risques de surdose. Le Paradote est un médicament sous forme de tablettes contenant 100 mg de méthionine et 500 mg de paracétamol. La méthionine est utilisée pour remplacer le glutathion et permet de protéger le foie en cas de surdose.
Vente
Le nom acétaminophène est utilisé aux États-Unis, au Canada, au Japon, en Corée du Sud, à Hong Kong et en Iran[159]. En Colombie et au Mexique on l'appelle « acetaminofén ». Dans les autres pays, on emploie le nom « paracétamol ».
Le paracétamol est le médicament le plus prescrit en France, et même la base des trois médicaments les plus prescrits (noms commerciaux : Doliprane, Dafalgan, Efferalgan), qui totalisent plus de 260 millions de doses[160].
Selon le rapport de 2005 de la Caisse nationale d'assurance maladie, la famille de médicaments la plus prescrite en France est celle des antalgiques, qui progresse encore de façon importante (+ 9,2 % par rapport à 2004) pour atteindre 340 millions de boîtes vendues[161] - [162].
On retrouve en tête de liste des dix médicaments les plus prescrits en quantité en France en 2005 trois antalgiques à base de paracétamol seul : le Doliprane (1er), l'Efferalgan (2e) et le Dafalgan (3e)[163]. Deux antalgiques avec du paracétamol associé se placent aussi dans les dix produits les plus prescrits : Propofan (6e) et Di-Antalvic (8e)[163].
Cinq des huit produits les plus prescrits en France sont des antalgiques contenant du paracétamol, avec une très forte progression pour certains comme le Doliprane (+15 %) et Dafalgan (+11 %). Selon un rapport de l'Agence française de sécurité sanitaire des produits de santé (AFSSAPS)[164], appelée Agence nationale de sécurité du médicament et des produits de santé (ANSM) depuis 2012, les prescriptions de paracétamol en France ont été multipliées par deux en dix ans. Cette forte croissance signifie que l'usage de ces antalgiques s'est banalisé, ce qui pourrait être dommageable à terme.
En termes de coût, le Doliprane qui est la spécialité la plus prescrite en quantité, ne se situe qu'au 15e rang des dépenses (96,3 millions d'euros, en progression de 11,7 % depuis 2004). L'Efferalgan est 42e avec 57,5 millions et +3,0 % et le Dafalgan 52e avec 47,5 millions et +7,9 %. L'ensemble des spécialités à base de paracétamol seul représente 236 millions d'euros (+12 % depuis 2004) et le 5e rang des dépenses[162].
Jusqu'en 2002, l'apparition de paracétamol générique en France a été bloquée pour sauvegarder l'emploi de l'usine Doliprane de Lisieux, dont les deux députés-maires successifs de 1953 à 1989 étaient pharmaciens. Le paracétamol était dans le domaine public mais son « généricage » était bloqué au prétexte de l'absence d'un dépôt de brevet spécifique[165]. Depuis, les ventes de paracétamol progressent plus vite que d'autres médicaments de la même classe (les antalgiques)[166], laissant supposer que certains prescripteurs et certains patients (auto-médication) ont changé leurs habitudes, passant de l'aspirine générique (ou de l'ibuprofène) au paracétamol générique.
On peut noter que dans le rapport de 2006 de la Caisse nationale d'assurance maladie les antalgiques conservent une croissance soutenue (+4 %) et restent en tête du classement des familles de médicaments les plus prescrites, avec 358 millions de boîtes prescrites et remboursées[167]. Ils demeurent les médicaments les plus prescrits, avec pour 2006, une croissance qui reste supérieure à +5 % pour Doliprane et Dafalgan[167]. Les antalgiques à base de paracétamol seul ou associé représentent 4 des 10 produits les plus prescrits et l'importante baisse du Propofan (-45,8 %) n'est que fictive quand on prend en compte le marché du groupe générique correspondant[167].
Le paracétamol fait partie de la liste modèle des médicaments essentiels de l'Organisation mondiale de la santé (liste mise à jour en )[168].
En 2015, les médicaments à base de cette molécule sont retirés des supermarchés suédois en raison de la hausse des intoxications depuis l'autorisation de leur vente en grandes surfaces en 2006[169].
En France, à compter du , le médicament n'est plus disponible en libre accès, mais doit être délivré par le pharmacien[170] (sans ordonnance).
Effets sur les animaux
Dans le cas d'une ingestion supposée pour les chats ou d'une surdose pour les chiens, il est important de consulter un vétérinaire immédiatement pour une désintoxication[171].
Le paracétamol est une substance extrêmement toxique pour les chats qui ne doivent en absorber dans aucun cas. Les chats ne possédant pas l'enzyme glucuronyl transferase, de petites quantités peuvent leur être fatales. La toxicité apparaît pour des doses journalières aussi faibles que 10 mg/kg[172]. Les symptômes initiaux sont le vomissement, la salivation et la décoloration de la langue et des gencives. Au bout de deux jours, les dommages corporels sont évidents et apparaît une jaunisse. Contrairement à ce qui se passe chez l'humain, ce ne sont pas les dommages hépatiques qui causent la mort mais c'est la production de méthémoglobine et de corps de Heinz dans les globules rouges qui empêche le transport de l'oxygène dans le sang, provoquant une mort par asphyxie. Des traitements efficaces sont possibles pour les faibles doses mais ils doivent être administrés très rapidement.
Pour les chiens, le paracétamol est un antalgique utile avec un bon résultat en matière d'efficacité, qui cause moins d'ulcères gastriques que les anti-inflammatoires non stéroïdiens. Mais il ne doit être administré que sur les conseils d'un vétérinaire. En effet, le surdosage, potentiellement mortel, est rapidement atteint même avec de faibles doses. La toxicité hépatique peut survenir à partir de 100 mg/kg et une méthémoglobinémie à partir de 200 mg/kg[172].
Le paracétamol est létal pour certains serpents et son utilisation dans le but de contrôler la prolifération du serpent brun arboricole (Boiga irregularis) dans l'île de Guam à l'aide de fausses souris imprégnées a été validée lors d'une étude[173].
Effets sur l'environnement
D'après une étude[174], le paracétamol pourrait se transformer en produit toxique, lorsque les usines de traitement des eaux usées utilisent le procédé de javellisation. Le paracétamol se transformerait, sous l'action de l'ion hypochlorite ClO−, en N-acétyl-p-benzoquinone imine et en 1,4-benzoquinone. La première molécule est toxique pour le foie tandis que la seconde est suspectée d'être génotoxique et mutagène. Des études supplémentaires doivent être effectuées pour savoir quelle est la concentration de ces substances à la sortie des eaux usées et pour connaître la persistance de ces produits dans l'environnement.
Fait divers
Le 30 septembre 1982, la première victime d'une série macabre meurt à Chicago après avoir absorbé une gélule d'acétaminophène (commercialisé sous le nom de Extra Strength Tylenol). Au total, sept personnes furent victimes de cet empoisonnement aux États-Unis[175] - [176]. Ces gélules contenaient du cyanure en quantité suffisamment importante pour être létale pour un adulte. La société Johnson & Johnson proposa alors d'échanger toutes les gélules de Tylenol en circulation par des comprimés de Tylenol. Dans cette affaire, la société eut une perte d'un million de dollars et fut condamnée à payer de lourdes indemnités aux victimes. Le responsable n'a jamais été arrêté et cette affaire n'a jamais été élucidée[177].
En février 1986, une nouvelle affaire d'empoisonnement éclate aux États-Unis, à la suite de la mort d'une jeune femme de 23 ans, Diane Elsroth, à Yonkers dans la région de New York, le . Elle avait absorbé une gélule de Tylenol Extra-Fort. La gélule avait été empoisonnée au cyanure, ce qui a relancé la psychose survenue après la vague de morts de Chicago, trois ans et demi auparavant[178]. À ce jour, tout comme pour la vague de crimes de 1982, cette affaire n'a pas été résolue.
Notes et références
Notes
- L'aspirine et l'ibuprofène sont des inhibiteurs des cyclooxygénases.
- Transformation en trois étapes, par réduction du NO2 en NH2 ; éthylation du groupe OH ; et finalement acylation du groupe NH2.
- En 1953, ils avaient déjà commercialisé l'Algoson, une préparation contenant du paracétamol et un sédatif.
- Des cas d'hépatites aiguës cytolytiques ont été rapportés à des doses thérapeutiques de paracétamol chez des sujets ayant une consommation chronique excessive d'alcool et donc un déficit en glutathion hépatique ce qui pourrait favoriser la survenue d'une nécrose hépatique sévère. Néanmoins cette notion est très controversée car la plupart de ces cas peuvent être dus à un surdosage non reconnu en paracétamol.
- Une consommation prolongée de paracétamol à des doses thérapeutiques pourrait provoquer une néphropathie chronique ; mais cette notion n'a jamais été confirmée par la suite.
- Hypotension parfois sévère, existante dans le cadre d'une réaction anaphylactique mais isolée, sans autres symptômes d'allergie.
Références
- « Details about INN Request 626 », sur extranet.who.int (consulté le )
- Entrée « 4-Acetamidophenol » dans la base de données de produits chimiques GESTIS de la IFA (organisme allemand responsable de la sécurité et de la santé au travail) (allemand, anglais), accès le 3 août 2018 (JavaScript nécessaire).
- Masse molaire calculée d’après « Atomic weights of the elements 2007 », sur www.chem.qmul.ac.uk.
- (en) « Paracétamol », sur ChemIDplus, (consulté le ).
- Fiche Sigma-Aldrich du composé Acetaminophen, consultée le 3 août 2018.
- « Acetaminophen », sur reciprocalnet.org (consulté le ).
- « Acétaminophène » dans la base de données de produits chimiques Reptox de la CSST (organisme québécois responsable de la sécurité et de la santé au travail), consulté le 25 avril 2009.
- IARC_Working_Group_on_the_Evaluation_of_Carcinogenic_Risks_to_Humans2009">IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, « Évaluations Globales de la Cancérogénicité pour l'Homme, Groupe 3 : Inclassables quant à leur cancérogénicité pour l'Homme », sur http://monographs.iarc.fr, CIRC, (consulté le ).
- PARACETAMOL, Fiches internationales de sécurité chimique .
- « Librairie de Molécules : Ibuprofène », sur PDB.
- ANSM, « Analyse des ventes de médicaments en France en 2013 » [PDF], sur ansm.sante.fr, .
- https://www.msdmanuals.com/fr/professional/blessures-empoisonnement/intoxications-empoisonnements/intoxication-par-le-paracétamol#:~:text=La%20N-acétylcystéïne%20est%20un,probablement%20par%20d'autres%20mécanismes.
- James J. Gormley, White willow bark is a gentle, effective pain-reliever, Better Nutrition, mars 1996, résumé (consulté le ).
- Merck index, 11e éd., 1989 (ISBN 0-911910-28-X). Morse, H. N. (1878), Ueber eine neue Darstellungsmethode der Acetylamidophenole. Berichte der deutschen chemischen Gesellschaft 11 (1) : 232–233. DOI 10.1002/cber.18780110151.
- Walter Sneader, Drug discovery : a history.
- Pain relief: from coal tar to paracetamol, RSC, . En ligne (consulté le ).
- Patrice Queneau, « La Saga du paracétamol », Médecine, vol. 2, no 4, , p. 158-159 (lire en ligne).
- Petite histoire du médicament.
- L'antipyrine (ou phenazone) a été inventée en 1883 par Ludwig Knorr comme substitut de la quinine (d'ailleurs Knorr a rattaché de manière erronée l'antipyrine à la famille chimique de la quinine). Knorr soumet sa découverte à Wilhelm Filehne (en) de l'université d'Erlangen, qui confirme ses propriétés antipyrétiques. L'antipyrine, utilisée en thérapeutique dès 1884, est le premier médicament de synthèse. L'antipyrine connaît un succès immédiat, notamment aux États-Unis (Janice Rae McTavish, Pain and Profits: the History of the Headache and its remedies in America). Knorr a déposé un brevet sur l'antipyrine — ce qui déroge aux usages de l'époque — et il a négocié une licence à un modeste producteur de colorants, Farbwerke Meister, Lucius und Brüning, qui deviendra la firme Hoechst et prendra le contrôle de Kalle and Co en 1908 ; le faible coût de l'acétanilide permet au médicament de perdurer malgré les risques attachés à sa consommation ; pour la même raison, il rentre illégalement dans la composition d'autres antipyrétiques. Cf. Walter Sneader, Drug Discovery: a History, p. 438 et (en) Milton Silverman, Mia Lydecker et Philip Randolph Lee, Bad Medicine : the Prescription Drug Industry in the Third World, Stanford, Stanford University Press, (ISBN 0-8047-1669-2), p. 87.
- La phénazone ou antipyrine a été largement utilisée pendant l'épidémie de grippe de 1918 (cf. Philippe Albou, Histoire du traitement de la fièvre avant l'aspirine, Lire en ligne).
- L'acétanilide, alors une substance d'utilisation commune dans le domaine de la teinturerie, ne pouvait faire l'objet d'un brevet : cet inconvénient fut contourné par la création du nom de marque Antifébrine : c'est d'ailleurs la première fois qu'un médicament a été commercialisé sous un nom de marque et non par son constituant chimique, et ce au grand dam des pharmaciens ; cf. Kevin M. Dunn, Caveman Chemistry : 28 Projects, from the Creation of Fire to the Production of Plastics, Universal-Publishers, 2003, p. 333 ; la pyramidone ou aminopyrine sera développée dans les années 1890 par Hoechst à partir de l'antifébrine.
- Mary Ellen Bowden, Amy Beth Crow, Tracy Sullivan, Pharmaceutical achievers: the human face of pharmaceutical research, p. 14.
- (en) Exp't 461 Acetaminophen, Tylenol [PDF], sur courses.chem.psu.edu.
- (en) Janice Rae McTavish, Pain and profits: the history of the headache and its remedies in America.
- François Chast, Histoire contemporaine des médicaments, édition La Découverte.
- Philippe Albou, Histoire du traitement de la fièvre avant l'aspirine, En ligne.
- (en) Mary Ellen Bowden, Amy Beth Crow, Tracy, Sullivan Pharmaceutical achievers: the human face of pharmaceutical research, p. 14.
- Ils ont publié une série de trois articles :
- (en) L. A. Greenberg, D. Lester, « The metabolic fate of acetanilid and other aniline derivatives, I. Major metabolites of acetanilid appearing in the urine », Journal of Pharmacology and Experimental Therapeutics 88 (1), 1946, p. 87–98, lire en ligne ;
- D. Lester, L. A. Greenberg, R.P. Carroll (1947), « The metabolic fate of acetanilid and other aniline derivatives, II. Major metabolites of acetanilid appearing in the blood », Journal of Pharmacology and Experimental Therapeutics 90 (1), 1947, p. 68–75, lire en ligne ;
- L. A. Greenberg, D. Lester, « The metabolic fate of acetanilid and other aniline derivatives, III. The role of p-aminophenol in the production of methemoglobinemia after acetanilid », Journal of Pharmacology and Experimental Therapeutics 90 (2), 1947, p. 150–153, lire en ligne.
- Ces quatre chercheurs font également partie de l'Institute for the Study of Analgesic and sedative drugs fondé en 1939 cf. (en) Mary Ellen Bowden, Amy Beth Crow, Tracy Sullivan in Pharmaceutical achievers: the human face of pharmaceutical research.
- (en) B. B. Brodie et J. Axelrod, « The fate of acetanilide in man », J. Pharmacol. Exp. Ther., vol. 94, no 1, , p. 29–38 (ISSN 0022-3565, résumé, lire en ligne).
- Brodie et Axelrod 1948, p. 37 « The analgesic action of acetanilide is exerted mainly through N-acetyl p-aminophenol which is an active analgesic ».
- Brodie et Axelrod 1948, p. 35 « The role of aniline in the formation of methemoglobin: Both methemoglobin and aniline were found in the blood of man after the administration of acetanilide. Methemoglobin was also found after the oral administration of aniline. This suggested that the methemoglobin in the blood after the ingestion of acetanilide might have been formed as a result of the aniline present. The nature of the actual methemoglobin-forming agent is not known. It has been considered to be p-aminophenol. This hypothesis is made unlikely since free p-aminophenol was not demonstrated in the blood after the administration of either acetanilide or aniline. It is possible that phenylhydroxylamine is the actual methemoglobin forming agent ».
- (en) The Julius Axelrod Papers - Work on the Sympathomimetic Amines, 1946-1958. Lire en ligne. « Axelrod and his mentor, Bernard Brodie, were charged with finding out why consumers who used non-aspirin analgesics, such as Bromo Seltzer, were developing an illness known as methemoglobinemia, a non-lethal blood condition. Brodie and Axelrod demonstrated that acetanilide, the main ingredient of these products, was the problem. They suggested that manufacturers replace it with acetaminophen ».
- (en) Milton Silverman, Mia Lydecker et Philip Randolph Lee, Bad Medicine : the Prescription Drug Industry in the Third World, Stanford, Stanford University Press, (ISBN 0-8047-1669-2), p. 89.
- (en) Walter Sneader, Drug discovery: a history, p. 439.
- « About | TYLENOL® », sur web.archive.org, (consulté le )
- (en) Aspirin adventures, a Festival of Analgesics. En ligne sur chemheritage.org, le site de la Chemical Heritage Foundation (consulté en ).
- Natasha Singer et Robert L. McNeil Jr., « Chemist Who Introduced Tylenol, Dies at 94 », The New York Times, (lire en ligne).
- (en) Mary Ellen Bowden, Amy Beth Crow, Tracy Sullivan, Pharmaceutical achievers: the human face of pharmaceutical research.
- (en) Site sur le médicament Panadol, consulté en .
- Patrice Queneau. Médecine. Volume 2, Numéro 4, 158-9, , Thérapeutiques. Lire en ligne P. Queneau donne 1961 comme date de commercialisation du Doliprane.
- Michel Pinaud, Benoît Vallet, V. Laudenbach, F. Kerbaul, Collectif, Conférences d'actualisation : 46e Congrès national d'anesthésie et de réanimation.
- (en) E. Maund, C. McDaid, S. Rice, K. Wright, B. Jenkins et N. Woolacott, « Paracetamol and selective and non-selective non-steroidal anti-inflammatory drugs for the reduction in morphine-related side-effects after major surgery: a systematic review », Br. J. Anaesth., vol. 106, no 3, , p. 292-297 (PMID 21285082, DOI 10.1093/bja/aeq406).
- (en) Paracetamol chemistry, sur pharmweb.net (consulté en ). En ligne.
- (en) Paracetamol manufacture review, sur pharmweb.net (consulté en ).
- (en) J. Bonnefont, J. P. Courade, A. Alloui et A. Eschalier, « Mechanism of the Antinociceptive Effect of Paracetamol », Drugs, vol. 63, no 2 (Spec), , p. 1-4 (ISSN 0012-6667 et 1179-1950, PMID 14758785, lire en ligne).
- « Actualité du paracétamol »(Archive.org • Wikiwix • Archive.is • Google • Que faire ?) (consulté le ), Évaluation et traitement de la douleur, 2006, p. 639-648, C. Remy, E. Marret, F. Bonnet, Département d'anesthésie-réanimation, hôpital Tenon, , Elsevier Masson SAS, (consulté le ).
- (en) Aronoff D.M., Oates J.A., Boutaud O., New insights into the mechanism of action of acetaminophen: Its clinical pharmacologic characteristics reflect its inhibition of the two prostaglandin H2 synthases, Clin. Pharmacol. Ther., 2006, 79 : 9-19, .
- Y a-t-il un rationnel à combiner le paracétamol et un AINS ? Kuntheavy-Roseline ING. En ligne [PDF].
- (en) COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: Cloning, structure, and expression N. V. Chandrasekharan, Hu Dai, K. Lamar Turepu Roos and col. .
- (en) Bonnefont J., Alloui A., Chapuy E. et al., Orally administered paracetamol does not act locally in the rat formalin test: evidence for a supraspinal, serotonin-dependent antinociceptive mechanism, Anesthesiology (en), 2003, 99, p. 976-981, .
- (en) Sprott H., Shen H., Gay S. et al., Acetaminophen may act through beta endorphin, Ann. Rheum. Dis., 2005, 64, 1522. En ligne.
- (en) The Organic Chemistry of Biological Pathways ; John McMurry et Tadhg Begley (en), p. 366 édition française.
- (en) « Peroxynitrite, the coupling product of nitric oxide and superoxide, activates prostaglandin biosynthesis » [PDF], Proc. Natl. Acad. Sci. USA, vol. 93, p. 15069–15074, .
- (en) Csaba Szabó, Harry Ischiropoulos et Rafael Radi, « Peroxynitrite: biochemistry, pathophysiology and development of therapeutics », Nat. Rev. Drug Discov., vol. 6, , p. 662-680 (DOI 10.1038/nrd2222).
- (en) « Nitric Oxide-Related Oxidative Stress and Redox Status » [PDF], Hindawi Publishing Corporation 2014 Article ID 129651.
- (en) « Acetaminophen inhibits prostanoid synthesis by scavenging the PGHS‐activator peroxynitrite », The Faseb Journal, 2007, vol. 22, no 1.
- H. Kupferschmidt. Traitement de l'intoxication au paracétamol. Centre Suisse d'Information Toxicologique. . « http://www.toxi.ch/upload/pdf/Merkblatt_Paracetamol_f.pdf »(Archive.org • Wikiwix • Archive.is • Google • Que faire ?) [PDF].
- Anti-inflammatoires non stéroïdiens et analgésie en période néonatale, R. Lenclen.
- Cours sur les antalgiques, Faculté de médecine, université Louis-Pasteur, Strasbourg, France, 2003. « En ligne »(Archive.org • Wikiwix • Archive.is • Google • Que faire ?) [PDF] (consulté le ).
- Développement pharmacologiques rationnel des associations d'analgésiques, Rev. Rhum. (éd. (fr)), 2003, 69, hors-série no 1. En ligne [PDF].
- C. Duru, P. Boudeville, M. Delalonde et N. Farah, « Masquage de goût du paracétamol par thermogranulation au palmitostéarate de glycérol », Ann. Pharm. Fr., vol. 62, no 3, , p. 186-192 (ISSN 0003-4509, DOI 10.1016/S0003-4509(04)94301-5, résumé).
- Médicaments à base de Paracétamol, Répertoire Commenté des Médicaments (Belgique).
- Paracétamol Ratiopharm, répertoire Vidal, (consulté le ).
- (en) E.M. Laska, A. Sunshine, I. Zighelboim et al., « Effect of caffeine on acetaminophen analgesia », Clin. Pharmacol. Ther., vol. 33, no 4, , p. 498-509 (lire en ligne [PDF]).
- (en) Diener H, Pfaffenrath V, Pageler L, Peil H, Aicher B, « The fixed combination of acetylsalicylic acid, paracetamol and caffeine is more effective than single substances and dual combination for the treatment of headache: a multicentre, randomized, double-blind, single-dose, placebo-controlled parallel group study. », Cephalalgia (en), vol. 25, no 10, , p. 776-787 (PMID 16162254) :
« the fixed combination of … caffeine was statistically significantly superior to the combination without caffeine. »
. - (en) E. Loder, « Fixed drug combinations for the acute treatment of migraine: place in therapy », CNS Drugs (en), vol. 19, no 9, , p. 769-84 (PMID 16142992) :
« benefits assumed for … caffeine … are not clearly confirmed in these trials. »
. - Patrice Queneau et Gérard Ostermann, Le médecin, le malade et la douleur, 4e éd., 2004, Masson (ISBN 2-294-01427-8). En ligne sur books.google.com.
- Dextropropoxyphène : toujours commercialisé, malgré les risques, Revue Prescrire, Numéro 288, . « la revue Prescrire déplore que la Commission française de la transparence (chargée de donner un avis sur le remboursement des médicaments) ait conclu que l'association dextropropoxyphène + paracétamol apportait un service médical rendu important, tout en disant ne pas disposer de comparaison de son effet antalgique par rapport au paracétamol seul ».
- BIAM, en substance : le Paracetamol, , sur biam.fr (consulté le ).
- (en) Prescott L.F., « Paracetamol: past, present, and future », Am. J. Ther. (en), vol. 7, no 2, , p. 143-7 (résumé).
- Les médicaments de la douleur, S Schück, H Allain. La douleur : moyens et stratégies thérapeutiques. La Revue du Praticien 1997 ; 47 : 555-69.
- Fièvre de l'enfant et du nourrisson, Dr H. Raybaud, sur esculape.com.
- Andrew Booth, Alan J. O'Rourke, À la recherche du niveau de preuve : théorie et pratique traduction d'un article paru dans Evidence-Based Medecine 1999;4(5):133-6 cf. : http://www.ebm-journal.presse.fr/numeros/24/805/index.php.
- (en) Heinz F. Eichenwald, « Fever and antipyresis », Bull World Health Organ, Genève, vol. 81, no 5, (lire en ligne).
- (en) Fiona M. Russell, Frank Shann, Nigel Curtis et Kim Mulholland, « Evidence on the use of paracetamol in febrile children », Bulletin of the World Health Organization, vol. 81, no 5, (lire en ligne [PDF]).
- (en) Janice E. Sullivan et Henry C. Farrar, « Clinical Report—Fever and Antipyretic Use in Children », Pediatrics (en), (DOI 10.1542/peds.2010-3852, lire en ligne [PDF]).
- Résumé des caractéristiques du produit, Paracétamol Sandoz 500 mg, gélule, .
- Recommandation AFSSAPS.
- Lettre d'information AFSSAPS.
- (en) « Acetaminophen Taken During Pregnancy Linked To Behavioral Problems In Children », (consulté le ).
- (en) Pre-natal exposure to paracetamol and risk of wheezing and asthma in children - A birth cohort study, Rebordosa et al., Int. J. Epidemiol. (en), .
- (en) Joanne E. Sordillo, Christina V. Scirica, Sheryl L. Rifas-Shiman et Matthew W. Gillman, « Prenatal and infant exposure to acetaminophen and ibuprofen and the risk for wheeze and asthma in children », The Journal of Allergy and Clinical Immunology, vol. 135, , p. 441–448 (ISSN 1097-6825, PMID 25441647, PMCID 4323723, DOI 10.1016/j.jaci.2014.07.065, lire en ligne, consulté le ).
- (en) Sander van den Driesche, Joni Macdonald, Richard A. Anderson et Zoe C. Johnston, « Prolonged exposure to acetaminophen reduces testosterone production by the human fetal testis in a xenograft model », Science Translational Medicine (en), vol. 7, , p. 288ra80 (ISSN 1946-6242, PMID 25995226, DOI 10.1126/scitranslmed.aaa4097, lire en ligne, consulté le ).
- (en) Morten Søndergaard Jensen, Cristina Rebordosa, Ane Marie Thulstrup et Gunnar Toft, « Maternal use of acetaminophen, ibuprofen, and acetylsalicylic acid during pregnancy and risk of cryptorchidism », Epidemiology (Cambridge, Mass.) (en), vol. 21, , p. 779–785 (ISSN 1531-5487, PMID 20805751, DOI 10.1097/EDE.0b013e3181f20bed, lire en ligne, consulté le ).
- David Møbjerg Kristensen, Ulla Hass et al., « Intrauterine exposure to mild analgesics is a risk factor for development of male reproductive disorders in human and rat », Human Reproduction (en), (lire en ligne).
- (en) Claudia A. Snijder, Andreas Kortenkamp, Eric A. P. Steegers et Vincent W. V. Jaddoe, « Intrauterine exposure to mild analgesics during pregnancy and the occurrence of cryptorchidism and hypospadia in the offspring: the Generation R Study », Human Reproduction (Oxford, England) (en), vol. 27, , p. 1191–1201 (ISSN 1460-2350, PMID 22301570, DOI 10.1093/humrep/der474).
- (en) K. Brandt, « [Paracetamol in the treatment of osteoarthritis pain] », Drugs, vol. 63 Spec No 2, , p. 23-41 (PMID 14758788).
- (en) Hansten P.D. et Horn J.R. in : Drug interactions. Analysis and management. A clinical perspective and analysis of current developments. Fact and comparisons. 2000.
- (en) Pickering, G, Loriot MA, Libert F, Eschalier A, Beaune P et Dubray C., « Analgesic effect of acetaminophen in humans: first evidence of a central serotonergic mechanism », Clin. Pharmacol. Ther., vol. 79, , p. 371-378 (DOI 10.1016/j.clpt.2005.12.307).
- (en) G.G. Graham, K.F. Scott et R.O. Day, « [Tolerability of paracetamol] », Drugs, vol. 63 Spec no 2, , p. 43-46 (PMID 14758789).
- (en) S.L. Mendizabal et M.L. Díez Gómez, « Paracetamol sensitivity without aspirin intolerance », Allergy (en), vol. 53, no 4, , p. 457-458 (PMID 9574897, lire en ligne [PDF]).
- (en) Y. Tsujino, N. Okamoto et E. Morita, « Acetaminophen-induced urticaria without aspirin intolerance », J. Dermatol. (en), vol. 34, no 3, , p. 224-226 (PMID 17291310, DOI 10.1111/j.1346-8138.2007.00257.x).
- (en) D. Bougie et R. Aster, « Immune thrombocytopenia resulting from sensitivity to metabolites of naproxen and acetaminophe », Blood, vol. 97, no 12, , p. 3846-3850 (PMID 11389025, lire en ligne).
- (en) S.L. Nuttall, J. Williams et M.J. Kendall, « Does paracetamol cause asthma? », J. Clin. Pharm. Ther. (en), vol. 28, no 4, , p. 251-257 (PMID 12911676, DOI 10.1046/j.1365-2710.2003.00492.x).
- (en) Paracetamol and asthma. Raghuram et Archer. Thorax (en). 2000 Oct;55(10):883; author reply 883-4. .
- (en) Treatment of pain or fever with paracetamol (acetaminophen) in the alcoholic patient: a systematic review. Dart et al., Am. J. Ther. (en), ; 7(2):123-34.
- (en) Paracetamol, alcohol and the liver, Prescott. Br. J. Clin. Pharmacol., avril 2000, 49(4):291-301. (en).
- (en) Acetaminophen-induced hypotension. Brown. Heart Lung (en). 1996 Mar-Apr;25(2):137-40. .
- (en) Anaphylactic shock induced by paracetamol, Van Diem et Grilliat. Eur. J. Clin. Pharmacol. (en) 1990, 38(4):389-90.
- (en) Acetaminophen (Paracetamol)-Induced Anaphylactic Shock. Bachmeyer et al. South Med J. (en) 2002 Jul;95(7):759-60. .
- (en) Vascular purpura caused by paracetamol. A case. Dussarat et al., Presse Med. 1988 Sep 17;17(31):1587. .
- (en) Medication use and the risk of Stevens-Johnson syndrome or toxic epidermal necrolysis. Roujeau et al. N. Engl. J. Med., décembre 1995, 14, 333(24):1600-7, .
- (en) Ano-rectal and colonic complications of suppositories and enemas. De Parades et al. Gastroenterol Clin Biol. 1996;20(5):446-52. . Note : lors de l'usage par voie rectale, souvent en association avec d'autres médicaments comme le dextropropoxyphène.
- (en) Agranulocytosis caused by paracetamol: a case, with positive readministration. Chichmanian et al. Ann Med Interne (Paris). 1989;140(4):332-3. .
- (en) Acute pancreatitis induced by codeine-acetaminophen association: report of two cases. Locher et al. Gastroenterol Clin Biol. 2003 Jan;27(1):124-5. .
- (en) Association of paracetamol and codeine, a rare cause of acute drug-induced pancreatitis. Casassus-Builhé et al. Presse Med. 2004 Apr 24;33(8):536. .
- (en) Does hepatitis due to subacute paracetamol toxicity exist? Apropos of 3 possible cases. Bidault et al. Thérapie (de). 1987 Jul-Aug ; 42(4):387-8. .
- (en) Paracetamol-induced cholestatic and granulomatous liver injuries. Lindgren et al., J. Intern Med. (en) 1997 May ; 241(5):435-9. .
- (en) Acetaminophen-induced rhabdomyolysis. Moneret-Vautrin et al. Allergy (en). 1999 Oct;54(10):1115-6. .
- Revue Médicale Suisse no 129.17/10/2007. Paracétamol : toxicité hépatique aux doses thérapeutiques et populations à risque. M. Seirafi A. Iten A. Hadengue .
- A. Mofredj, J.-F. Cadranel, B. Darchy, J.-C. Barbare, A. Cazier, V. Pras et M. Biour, « Toxicité hépatique du paracétamol à dose thérapeutique chez le sujet éthylique chronique », Annales de médecine interne, vol. 150, no 6, , p. 507-511.
- (en) Beasley R., Clayton T., Crane J. et al., Association between paracetamol use in infancy and childhood, and risk of asthma, rhinoconjunctivitis, and eczema in children aged 6–7 years: analysis from Phase Three of the ISAAC programme, Lancet, 2008 ; 372:1039-1048.
- (en) Amberbir A. et al., The role of acetaminophen and geohelminth infection on the incidence of wheeze and eczema. A longitudinal birth-cohort study, Am. J. Respir. Crit. Care Med. (en), 2011 ; 183 : 165-170.
- MacIntyre IM, Turtle EJ, Farrah TE et al. Regular acetaminophen use and blood pressure in people with hypertension: the PATH-BP trial, Circulation, 2022;145:416–423
- (en) Fort DJ, Rayburn JR, Bantle JA, Evaluation of acetaminophen-induced developmental toxicity using FETAX, Drug Chem Toxicol (en), 1992;15329-50.
- Katrin Faber, « Intoxication aiguë au paracétamol », Forum Médical Suisse, no 38, , p. 647-651 (lire en ligne).
- « La mention « surdosage = danger » sera obligatoire sur les boîtes de paracétamol », Le Monde, (lire en ligne, consulté le ).
- (en) Dart R.C., Erdman A.R., Olson K.R., Christianson G., Manoguerra A.S., Chyka P.A., Caravati E.M., Wax P.M., Keyes D.C., Woolf A.D., Scharman E.J., Booze L.L. et Troutman W.G., American Association of Poison Control Centers (en), « Acetaminophen poisoning: an evidence-based consensus guideline for out-of- hospital management », Clinical Toxicology, vol. 44, no 1, , p. 1–18 (PMID 16496488, résumé).
- V. Danel, « Intoxication aiguë par le paracétamol », Carli P. Protocoles 2004 - Urgences, plans et schémas thérapeutiques, Éditions Scientifiques, L, 2004, p. 193-195.
- Bernard Calvino (ESPCI-CNRS), « Paracétamol : ne pas dépasser la dose prescrite », Pour la science no 385, .
- « Même faible, le surdosage de paracétamol est dangereux », sur sante.lefigaro.fr, Le Figaro, (consulté le ).
- (en) Hepatotoxicity caused by therapeutic doses of paracetamol in alcoholics. Report of 2 cases of fatal hepatitis in cirrhosis, Mofredj, Cadranel J.F., Darchy B., Barbare J.C., Cazier A., Pras V., Biour M., Annales de médecine interne (Paris). 1999 Oct ; 150(6):507-11.
- (en) M.D. McGoldrick et G.R. Bailie, « Nonnarcotic analgesics : Prevalence and estimated economic impact of toxicities », The Annals of Pharmacotherapy (en), vol. 31, no 2, , p. 221-227 (PMID 9034424).
- (en) Glutathione synthetase-deficient lymphocytes and acetaminophen toxicity. Spielberg SP, Gordon GB. Clin. Pharmacol. Ther., janvier 1981, 29(1):51-5, DOI 10.1038/clpt.1981.9.
- (en) O. Moling, E. Cairon, G. Rimenti, F. Rizza, R. Pristerá et P. Mian, « Severe hepatotoxicity after therapeutic doses of acetaminophen. », Clin Ther. (en), vol. 28, no 5, , p. 755-760 (PMID 16861097, DOI 10.1016/j.clinthera.2006.05.002).
- (en) B.H. Lauterburg, « Analgesics and glutathione », American Journal of Therapeutics (en), vol. 9, no 3, , p. 225-233 (résumé).
- https://www.leparisien.fr/societe/sante/naomi-musenga-victime-d-une-intoxication-au-paracetamol-11-07-2018-7816884.php
- (en) « Paracetamol: How Much To Take & What an Overdose Looks Like », sur pharmweb.net (consulté le ).
- (en) Tenenbein M, « Acetaminophen: the 150 mg/kg myth », J. Toxicol. Clin. Toxicol., vol. 42, no 2, , p. 145–148 (PMID 15214618, résumé).
- M. Grégoire et al., « L’acidose lactique précoce lors de l’intoxication massive au paracétamol : un trouble métabolique parfois méconnu », Toxicologie analytique et clinique, Elsevier, vol. 26, no 2, (DOI 10.1016/S2352-0078(14)70086-1, présentation en ligne).
- « DOLIPRANE 1000 mg gél », sur VIDAL (consulté le )
- Lejonc JL, Elkharrat D, Lapandry C, Leblanc JP, Robert R, Saint-Martin J et al. Épuration digestive lors des intoxications aiguës. Réan Urg 1993 ; 2 (2 bis) : 169-75.
- (en) Krenzelok E.P., McGuigan M., Lheureux P., Position statement: Ipecac syrup., J. Toxicol. Clin. Toxicol., 1997 ; 35 : 699-709.
- (en) Vale J.A., Kulig K., American Academy of Clinical Toxicology, European Association of Poisons Centres and Clinical Toxicologists, « Position paper: gastric lavage », J. Toxicol. Clin. Toxicol., vol. 42, no 7, , p. 933–43 (PMID 15641639, résumé).
- (en) Ekins B, Ford D, Thompson M, Bridges R, Rollins D, Jenkins R, « The effect of activated charcoal on N-acetylcysteine absorption in normal subjects », Am. J. Emerg. Med. (en), vol. 5, no 6, , p. 483–487 (PMID 3663288).
- Intoxication au paracétamol, 2003, sur urgences-serveur.fr.
- N-acétylcystéine, 2004, sur urgences-serveur.fr.
- (en) Clayden, Greeves, Warren, Wothers, Organic Chemistry, p. 1357.
- « Paracetamol Information Centre - Paracetamol - Metabolism, Biochemistry of Overdose and its Treatment », sur pharmweb.net (consulté le ).
- V. Danel et P. Saviuc, Syndrome hépatotoxique, Médecine d'urgence, 2005, p. 41-50. « En ligne »(Archive.org • Wikiwix • Archive.is • Google • Que faire ?) (consulté le ) sur anesthesie-foch.org.
- (en) Prescott LF, Illingworth RN, Critchley JA, Stewart MJ, Adam RD, Proudfoot AT: Intavenous N.acetylcysteine - the treatment of choice for paracetamol poisoning. Br Med J ii: 1097-1100, 1979. .
- (en) Smilkstein M.J., Bronstein A.C., Linden C., Augenstein W.L., Kulig K.W. et Rumack B.H., Acetaminophen overdose - a 48-hour intravenous N-acetylcysteine treatment protocol, Ann. Emerg. Med. (en), 20, 1058-63, 1991. Résumé.
- (en) Smilkstein M.J., Knapp G.L., Kulig K.W., Rumack B.H., Efficacy of oral N.acetylcysteine in the treatment of acetaminophen overdose, New Engl. J. Med., 319, 1557-62, 1988, .
- (en) Rumack B.H., Peterson RG: Acetaminophen overdose - incidence, diagnosis, and mangement in 416 patients. Pediatrics (en) 62(5 pt 2 suppl.): 898-903, 1978. Résumé.
- Pharmacologie des anti-inflammatoires non-stéroïdiens et pathologies ORL. En ligne.
- (en) C. J. Nikles, M. Yelland, C. Del Mar et D. Wilkinson, « The Role of Paracetamol in Chronic Pain : An Evidence-Based Approach », American Journal of Therapeutics (en), vol. 12, , p. 80-91 (présentation en ligne).
- (en) García Rodríguez LA, Hernández-Díaz S, « The risk of upper gastrointestinal complications associated with nonsteroidal anti-inflammatory drugs, glucocorticoids, acetaminophen, and combinations of these agents », Arthritis Research and Therapy (en), (PMID 11178116, résumé, lire en ligne).
- (en) Alano M.A., Ngougmna E., Ostrea E.M. Jr. et al., « Analysis of nonsteroidal anti-inflammatory drugs in meconium and its relation to persistent pulmonary hypertension of the newborn », Pediatrics (en), vol. 107, no 3, , p. 519-523 (PMID 11230592, résumé, lire en ligne).
- (en) Clark E, Plint AC, Correll R, Gaboury I, Passi B, « A randomized, controlled trial of acetaminophen, ibuprofen, and codeine for acute pain relief in children with musculoskeletal trauma », Pediatrics (en), vol. 119, no 3, , p. 460-467 (PMID 17332198, résumé).
- (en) Goldman R.D., Ko K., Linett L.J., Scolnik D., « Antipyretic efficacy and safety of ibuprofen and acetaminophen in children », Ann Pharmacother (en), vol. 38, no 1, , p. 146-150 (résumé).
- (en) Perrott D.A., Piira T., Goodenough B., Champion G.D., « Efficacy and safety of acetaminophen vs ibuprofen for treating children's pain or fever: a meta-analysis », Arch Pediatr Adolesc Med (en), vol. 158, no 6, , p. 521-526 (PMID 15184213, résumé).
- (en) Kramer M.S., Naimark L.E., Roberts-Brauer R. et al., « Risks and benefits of paracetamol antipyresis in young children with fever of presumed viral origin », Lancet, vol. 337, no 8741, , p. 591-594 (résumé).
- « Fièvre chez l'enfant », Recommandations AFSSAPS et ANAES, , sur esculape.com.
- (en) Janice Tanne, « Paracetamol causes most liver failure in UK and US », British Medical Journal, vol. 332, , p. 628 (résumé).
- (en) Safety issues related to acetaminophen, Food and Drud Administration, Center for drug evaluation and research, non prescription drugs advisory committee, september 19, 2002. « Annually there were over 56 000 emergency department visits, more than 26 000 hospitalizations, and 458 deaths associated with APAP. These numbers represent both intentional and unintentional overdoses. » - « This slide represents the number of estimated cases of unintentional overdoses. Again, these groupings are independent from each other and represent annual averages. There were over 13 000 emergency department visits, more than 2 000 hospitalization, and 100 deaths associated with APAP » - « In 1995, overall APAP related fatalities were at least 76, and this increased dramatically to 141 in 1991. APAP is the leading pharmaceutical agent associated with deaths in tests and represented about 60 percent of all deaths that were reported to tests in 1999. This slide gives a breakdown of the intentionality among 141 APAP related fatalities in 1999. Sixty-five percent of the cases were suicidal and 30 percent of the cases were unintentional ».
- « Antidouleurs : gare à la surconsommation ! », sur doctissimo.fr.
- F. Adnet, S. Atout, M. Galinski et F. Lapostolle, « Évolution des intoxications médicamenteuses volontaires en France », Réanimation, vol. 14, no 8, , p. 721-726 (ISSN 1624-0693, résumé).
- (en) Bradley, N., « BMJ should use « paracetamol » instead of « acetaminophen » in its index », BMJ, vol. 313, no 7058, , p. 689 (PMID 8811774, PMCID 2351967).
- Le Moniteur des pharmacies, cahier III, no 2634, .
- Les 10 médicaments les plus prescrits en 2005 - Les antalgiques en tête du classement, sur agevillage.com.
- Médicaments remboursables : analyse des principales évolutions de l'année 2005 [PDF], l'Assurance maladie, Point d'information mensuel, .
- AFFSAPS, Les ventes de médicaments aux officines et aux hôpitaux en France - Chiffres-clés 2005, (lire en ligne)
- Analyse des ventes de médicaments aux officines et aux hôpitaux en France 1995-2005, 7e édition, , disponible en ligne [PDF]. Voir le tableau p. 107.
- Inscription du paracétamol sur le registre des génériques, Sénat (France), 2002.
- « Première analyse sur 10 ans de l’évolution de l’utilisation des antalgiques en France », sur VIDAL (consulté le )
- Médicaments remboursables : analyse des principales évolutions de l'année 2006 [PDF], Caisse nationale de l'Assurance Maladie - .
- (en) « WHO Model List of Essential Medicines, 18th list » [PDF], .
- France Soir, « Le paracétamol retiré des grandes surfaces en Suède », sur francesoir.fr, (consulté le ).
- Point d'Information ANSM - Bon usage du paracétamol et des anti-inflammatoires non stéroïdiens (AINS) : ces médicaments ne pourront plus être présentés en libre accès.
- (en) Villar D., Buck W.B., Gonzalez J.M., « Ibuprofen, aspirin and acetaminophen toxicosis and treatment in dogs and cats », Vet Hum Toxicol, vol. 40, no 3, , p. 156-62 (PMID 9610496, résumé).
- Intérêt et risques d'utilisation du paracétamol chez les animaux de compagnie. Folia veterinaria. En ligne [PDF], sur bcfi-vet.be.
- (en) Johnston J, Savarie P, Primus T, Eisemann J, Hurley J, Kohler D, « Risk assessment of an acetaminophen baiting program for chemical control of brown tree snakes on Guam: evaluation of baits, snake residues, and potential primary and secondary hazards », Environ Sci Technol, vol. 36, no 17, , p. 3827–33 (PMID 12322757, lire en ligne) :
« The brown tree snake (Boiga irregularis) is a significant ecological, agricultural, and economic pest on Guam. Acetaminophen has recently been identified as a promising snake toxicant. Subsequent experimentation has shown that acetaminophen-mouse baits are readily consumed by and acutely toxic to brown tree snakes. »
. - Environmental Science & Technology (ISSN 0013-936X), Environmental Science and Technology, 2006, vol. 40, no 2, p. 516-522.
- (en) Tylenol molecule, sur worldofmolecules.com.
- (en) Andrew H. Malcom, 100 Agents hunt for killer in 7 tylenol deaths, The New York Times, , sur query.nytimes.com (consulté le 23 décembre 2007).
- (en) I. Wilkerson, Tylenol Maker Settles in Tampering Deaths, The New York Times, , sur query.nytimes.com (consulté le 23 décembre 2007).
- (en) Spencer Davidson, A Replay of the Tylenol Scare, Time, .
Annexes
Bibliographie
- C. Remy, E. Marret, F. Bonnet, « Actualité du paracétamol »(Archive.org • Wikiwix • Archive.is • Google • Que faire ?) (consulté le ), Évaluation et traitement de la douleur, 2006, p. 639-648, Département d'anesthésie-réanimation, hôpital Tenon, 9 septembre 2006, Elsevier Masson SAS.
- (en) Paracetamol Information Centre : « an independent source of information, fact, and background detail, for journalists on all aspects of the clinical and home use of paracetamol ».
Articles connexes
Liens externes
- Compendium suisse des médicaments : spécialités contenant Paracétamol
- Résumé