Adsorption
En chimie, l’adsorption est un phénomène de surface par lequel des atomes, des ions ou des molécules - des adsorbats - se fixent sur une surface solide - l'adsorbant - depuis une phase gazeuse, liquide ou une solution solide[1]. Dans le cas d'un atome adsorbé, on parle d'adatome. Ce phénomène est différent de l'absorption, par lequel un fluide ou le composant d'une solution solide rentre dans le volume d'une autre phase liquide ou solide, mais les deux effets sont similaires et sont facilement (et à tort) confondus, notamment dans des applications pour le grand public[2]. Un exemple usuel typique est la fixation de vapeur d'eau sur une vitre. Industriellement, l'adsorption s'effectue à l'intérieur de grains d'adsorbant, sur la surface développée par les pores afin d'obtenir une grande quantité fixée dans un minimum de volume.
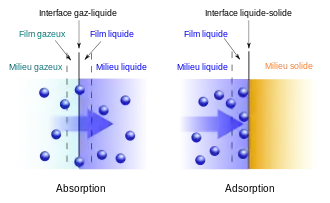
Le processus d'adsorption est donc basé sur l'interaction de l'adsorbat avec une surface, ce qui peut faire intervenir divers processus plus ou moins intenses comme les interactions de Van der Waals, les interactions dipolaires, ou les liaisons chimiques covalentes ou ioniques[3].
Le phénomène inverse, par lequel les molécules adsorbées sur une surface s’en détachent, notamment sous l’action de l’élévation de la température, ou de la baisse de pression, se nomme la désorption.
Ce phénomène a une très grande importance dans de nombreux processus physiques et chimiques[4] : capture de polluants[5], séparation de gaz, catalyse, etc. Il est aussi la base de nombreuses méthodes de caractérisation des solides comme la mesure des surfaces spécifiques ou l'étude de la porosité[6]. En mécanique industrielle, il joue un rôle fondamental dans les processus de lubrification et les procédés de brasage.
Le terme « adsorption » fut utilisé pour la première fois en 1881 par le physicien allemand Heinrich Kayser[7]. L'étude de l'adsorption est basée sur la mesure de la corrélation entre la concentration d'adsorbat dans la phase fluide et la quantité d'adsorbat qui est piégée par la surface à une température donnée : c'est la mesure des isothermes d'adsorption[3] - [4]. De très nombreux modèles ont été et sont toujours créés pour rendre compte des mesures expérimentales, certains sont très simples (isotherme de Langmuir ou de Freundlich) alors que d'autres nécessitent des calculs complexes (simulations basées sur la théorie de la fonctionnelle de la densité ou sur la méthode Monte-Carlo).
Historique
Le phénomène d'adsorption était utilisé depuis très longtemps dans la vie pratique, principalement par l'usage du charbon actif dans des applications médicales ou pour la purification d'eau. Il a cependant fallu attendre la fin du XVIIIe siècle pour que l'on commence à étudier la capture par un solide d'une espèce en phase gazeuse[8], puis d'un colorant en solution aqueuse[9]. De Saussure observa ensuite l'aspect exothermique de ce phénomène[10].
Les premières applications industrielles furent liées à l'adsorption sélective permettant la séparation de composés gazeux ou liquides, ouvrant la voie à de nombreuses applications telles que la purification d'eau ou d'air. La première analyse théorique de l'adsorption fut due à Irving Langmuir en 1914, elle décrivait l'adsorption d'une monocouche d'adsorbat sur une surface homogène sous la forme d'une équation, l'isotherme de Langmuir.
De très nombreux développements ont été publiés durant tout le XXe siècle pour prendre en compte l'adsorption multicouche sur des surfaces complexes [11]. L'isotherme d'adsorption le plus utilisé est celui de Brunauer, Emmet et Teller [12], cette équation BET généralise l'approche de langmuir à une adsorption multicouche et est devenue le modèle standard pour la détermination de la surface spécifique des solides [6]. Les trois dernières décennies ont vu se développer de nombreux modèles utilisant la théorie de la fonctionnelle de la densité ou la méthode Monte-Carlo qui sont basées sur une description des interactions entre l'adsorbat et l'adsorbant à l'échelle moléculaire.
Aspects théoriques
Types d'interactions
Selon la nature des interactions qui retiennent l'adsorbat sur la surface de l'adsorbant, l'adsorption peut être classée en deux familles :
- l’adsorption physique ou physisorption met en jeu des liaisons faibles, du type forces de van der Waals entre les espèces chimiques adsorbées et l’adsorbant. Ces liaisons sont analogues à celles qui sont impliquées lors d’une liquéfaction. Elle se produit cependant bien avant que le gaz n’atteigne une pression égale à sa pression de vapeur saturante, à des températures assez basses et voisines du point d’ébullition de la phase adsorbée. Elle est en général réversible et l’équilibre est obtenu lorsque les vitesses d’adsorption et de désorption sont égales. L’adsorption physique est favorisée par une baisse de la température ;
- l'adsorption chimique ou chimisorption met en jeu des énergies de liaison importantes, du type liaisons covalentes, ioniques ou métalliques entre les espèces chimiques adsorbées et l’adsorbant. Elle s’accompagne d’une profonde modification de la répartition des charges électroniques des molécules adsorbées. Elle est souvent irréversible (ou difficilement réversible). Comme elle nécessite la formation d'interactions de forte énergie et à courte distance, la chimisorption engendre la formation d'une couche monomoléculaire.
Le premier cas correspond par exemple à l'adsorption de diazote sur un matériau poreux[3], alors que le second est observé lors de l'adsorption de dioxygène sur un matériau carboné à 300 °C[13]. Dans le cas où l'adsorbat est en solution dans un solvant, les interactions électrostatiques peuvent jouer un rôle très important. Dans ce cas, l'isotherme d'adsorption sera fortement influencé par le pH de la solution car il peut modifier la charge de surface de l'adsorbant et la polarité des espèces en solution. C'est par exemple le cas pour l'adsorption sur des matériaux carbonés de colorants[14] ou de polluants comme les composés phénoliques[15].
Phase gazeuse
Pour une température , l'équation caractéristique décrivant le phénomène d'adsorption depuis la phase gazeuse est la quantité d'adsorbat capturée par la surface de l'adsorbant en fonction de la pression d'adsorbat :


étant le nombre de moles adsorbées et la masse d'adsorbant. Dans le cas où le fluide n'est pas en état supercritique, on utilise souvent la pression relative , étant la pression de vapeur saturante à la température . Cet isotherme peut être complexe car plusieurs processus peuvent être mis en jeu dans l'adsorption. De façon générale, on peut indiquer trois grandes régions :




- faible pression : l'adsorption nécessite une forte affinité de l'adsorbat avec la surface, ce qui est souvent observé lors de l'adsorption dans les pores de taille nanométriques ;
- moyenne pression : l'adsorption peut se faire même si l'énergie d'interaction est plus modérée, on observe donc en général l'adsorption par condensation capillaire dans les pores de 5 à 50 nm ;
- forte pression : on approche de la pression de vapeur saturante, donc on commence à observer de la condensation entre les grains du matériau.
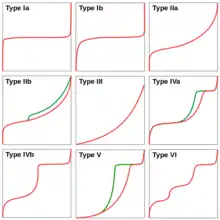
Cinq formes caractéristiques ont été définies par Brunauer et al.[16]. Cette classification a ensuite été affinée par le groupe de travail sur l'adsorption de l'IUPAC[3] - [4]. On constate que dans certains cas les isothermes d'adsorption sont différents des isothermes de désorption, le phénomène d'adsorption n'est donc pas toujours parfaitement réversible. Ces isothermes correspondent aux adsorbants suivants :
- type Ia et Ib : observés pour des adsorbants microporeux, c'est-à-dire des matériaux pour lesquels la porosité est principalement constituée de pores de moins de 2 nanomètres ;
- type IIa et IIb : correspond à des agrégats de grains non poreux, par exemple des argiles, des ciments, des pigments ;
- type III : adsorption de très faible énergie sur un échantillon non poreux, c'est un cas rarement observé ;
- type IV : ces isothermes correspondent à des matériaux mésoporeux, c'est-à-dire des solides pour lesquels le volume poreux est principalement constitué de pores ayant une taille comprise entre 2 et 50 nm ;
- type V : est similaire au type IV, mais indique une faible énergie d'interaction entre l'adsorbat et l'adsorbant ;
- type VI : est un isotherme à marches, il indique la formation successive de couches adsorbées sur la surface de l'adsorbant.
Phase liquide
Si l'adsorbat est une molécule en solution dans un solvant, l'équation caractéristique devient :

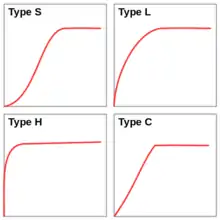
Les formes caractéristiques des isothermes ont été classifiées par Giles[17] en quatre groupes principaux:
- type S : correspond à une adsorption dans laquelle interviennent les interactions adsorbat-adsorbant, mais aussi adsorbat-adsorbat. On peut donc observer une adsorption coopérative de molécules ;
- type L : forme correspondant à l'isotherme de Langmuir ;
- type H : correspond à une forte affinité entre l'adsorbat et l'adsorbant, ce qui conduit à une adsorption importante même si la concentration est faible ;
- type C : correspond à un isotherme linéaire, ce qui veut dire que le nombre de nouveaux sites d'adsorption sont créés lors de l'adsorption.

Des sous-groupes ont été définis pour rendre compte de la diversité des mesures expérimentales[18].
Mesure des isothermes d'adsorption
Il existe de nombreuses méthodes permettant de déterminer les courbes caractéristiques de l'adsorption[3] - [6] - [19]. les points importants sont les suivants :
- il faut mesurer deux paramètres à l'équilibre : la quantité adsorbée et la concentration ou la pression partielle d'adsorbat ;
- il faut être à l'équilibre, ce qui peut réclamer un temps conséquent : plusieurs heures en phase gazeuse et plusieurs jours en phase liquide. Ceci est souvent lié à la faible taille de pore des adsorbants, ce qui conduit à une réduction très importante de la vitesse de diffusion des molécules d'adsorbat vers la surface du solide [20] ;
- il faut contrôler précisément la température lors des mesures.
Les méthodes diffèrent suivant que l'on étudie l'adsorption en phase gazeuse ou en solution.
Manométrie
Cette méthode est utilisée pour la phase gazeuse. La première étape est de nettoyer l'échantillon en enlevant tout ce qui peut être adsorbé sur sa surface, cette étape appelée dégazage se fait en mettant l'échantillon sous un vide poussé (10-6 mmHg) avec le cas échéant un chauffage modéré de l'échantillon. La mesure proprement dite s'effectue comme suit :
- l'échantillon de volume est mis dans une cellule de volume sous vide et un autre volume est rempli avec une pression d'adsorbat ;
- on ouvre la vanne entre les deux cellules et on laisse la pression s'équilibrer, s'il n'y aucune adsorption la pression finale sera , s'il y a adsorption la pression sera et la quantité de matière adsorbée est proportionnelle à la différence de pression observée ;
- pour faire le point suivant, on recommence avec une pression plus élevée.






Cette méthode a l'avantage important de permettre le calcul des deux grandeurs désirées avec une seule mesure de pression. Elle est aussi facilement automatisable et plusieurs sociétés proposent des appareillages totalement automatiques[21].
Isothermes d'adsorption en solution
Le principe est de mettre en contact une masse d'adsorbant avec un volume d'une solution d'adsorbat à la concentration initiale , l'adsorption va provoquer une diminution de cette concentration. Lorsque l'équilibre est atteint, il faut donc séparer le solide de la solution libre (que l'on nomme surnageant), et mesurer sa concentration en adsorbat . La quantité adsorbée est .




Comme indiqué plus haut, il est en général nécessaire de faire au préalable une étude cinétique de l'adsorption pour établir le temps nécessaire à l'établissement de l'équilibre. D'autre part, il existe un effet de matrice puisque les autres espèces présentes dans le milieu peuvent aussi s'adsorber sur le solide, on étudie donc la compétition entre les différents adsorbats présents.
Interprétation des isothermes d'adsorption
Physisorption
L'objectif est de corréler les isothermes d'adsorption avec des propriétés caractéristiques de l'adsorbant et de l'adsorbat :
- nature de l'interaction, quantification de l'énergie d'interaction ;
- propriétés de la phase adsorbée: densité, arrangement des molécules ;
- texture de l'adsorbat: surface spécifique, volume poreux, taille et forme des pores.
De très nombreuses équations et modèles ont été créés depuis le début du XXe siècle pour accéder à ces informations [3] - [11]. La difficulté est que pour un matériau donné, il peut y avoir différents types de phénomènes qui interviennent. Les différentes approches possibles sont les suivantes :
- la comparaison avec des isothermes de référence obtenus sur des matériaux modèles ;
- la simplification à un type d'interaction, ce qui permet de ramener tout ou partie de l'isotherme à un seul phénomène physique ;
Depuis la fin du XXe siècle, une autre approche consiste à construire un modèle atomique du matériau et à calculer l'isotherme que l'on devrait obtenir. La comparaison avec les isothermes expérimentaux permet de valider le modèle[22].
Isotherme de Freundlich
Il existe de nombreuses possibilités pour ajuster des données expérimentales avec une équation mathématique. Dans le domaine de l'adsorption, l'équation la plus utilisée est celle de Freundlich[23]:

étant une constante supérieure à 1. La complexité des isothermes obtenus expérimentalement s'accorde cependant rarement avec cette simple loi de puissance sur un large domaine de pression. De plus, cette équation ne tend pas vers la Loi de Henry lorsque la pression tend vers 0.


Loi de Henry
La Loi de Henry, initialement définie pour étudier la quantité de gaz dissout dans un liquide, recouvre un principe plus général que l'on peut appliquer à l'adsorption : lorsque la concentration en adsorbat tend vers 0, la quantité adsorbée tend aussi vers 0. De plus, la relation entre quantité adsorbée et concentration en adsorbat est linéaire :

Tout modèle d'adsorption devrait donc se ramener à cette équation lorsque tend vers 0. Sur le plan pratique, cette équation est cependant peu utilisée car la surface des matériaux est hétérogène et comporte différents sites d'adsorption, donc même en supposant que l'adsorption sur ces différents sites se fait de façon indépendante, il y a donc autant de valeurs de que de types de sites d'adsorption. Toutefois, c'est la loi sur laquelle repose la chromatographie analytique: chaque constituant du mélange à sa propre valeur de k, et, à cause de la linéarité, chaque constituant s'adsorbe indépendamment des autres. Le chromatogramme d'un mélange est donc identique à la superposition des chromatogrammes de chaque constituant injecté pur. ceci permet de faire un étalonnage avec des corps purs.

Modèle de Langmuir
C'est le plus ancien modèle tentant de décrire le phénomène d'adsorption sur une surface plane homogène [24]. Il conduit à l'équation:

dans laquelle est la quantité maximale de molécule nécessaire à la formation d'une monocouche sur la surface, et est une constante caractéristique de l'affinité de l'adsorbat avec la surface. Cet isotherme est principalement utilisé lorsque l'adsorption conduit à la formation d'une monocouche, caractéristique d'une interaction forte entre la surface et l'adsorbat. Ce que l'on observe par exemple en catalyse. On constate que cette équation tend vers la loi de Henry lorsque la pression tend vers 0.


Cette équation a donné lieu à un certain nombre de variantes, la plus connue est l'équation de Langmuir-Freundlich (aussi nommée équation de Sips) qui combine l'équation empirique de Freundlich avec le modèle de Langmuir [25]:

Modèle BET
Ce modèle a été développé par Stephen Brunauer, Paul Hugh Emmett et Edward Teller en 1938[12]. C'est une extension du modèle de Langmuir à une adsorption en multicouches sur une surface plane homogène, on aboutit à l'équation suivante :

ou en utilisant la pression relative :

est la constante BET qui est caractéristique de l'interaction entre l'adsorbat et l'adsorbant.

Cette théorie est la base de la méthode standard pour la mesure de la surface spécifique, nommée surface BET par référence aux initiales des auteurs[6]. Ce modèle est principalement adapté aux matériaux comportant des pores ayant une taille supérieure à quelques nanomètres. Il est cependant aussi utilisé pour les matériaux microporeux (taille de pore inférieure à 2 nanomètres), la surface obtenue dans ce cas est alors plutôt un index caractéristique du matériau qu'une vraie valeur de la surface spécifique[26].
Modèle BJH
Ce modèle est basé sur la condensation capillaire dans un milieu poreux, donc sur l'équation de Kelvin. Il a été formalisé par E. P. Barrett, L. G. Joyner et P. P. Halenda en 1951 [27]. Le principe est de considérer que la porosité du matériau est constituée d'un ensemble de pores cylindriques indépendants les uns des autres. Pour une pression de vapeur donnée, on observera une condensation dans tous les pores dont le rayon est inférieur au rayon de Kelvin de la vapeur :


dans laquelle est la pression de vapeur saturante, est la tension de surface, est le volume molaire du liquide, est la Constante universelle des gaz parfaits et est la température. Par dérivation de l'isotherme d'adsorption, on peut donc calculer la distribution en taille des pores du matériau. Étant basé sur l'équation de Kelvin, il s'applique dans un domaine de validité de cette équation qui correspond aux pores dont la taille est supérieure à quelques nanomètres, c'est-à-dire le domaine des mésopores. Cette méthode est encore la plus utilisée pour l'étude de la mésoporosité des matériaux [6].




Isotherme d'adsorption sur une surface hétérogène
Un adsorbant peut contenir différents types de sites d'adsorption mettant en jeu des phénomènes physiques et chimiques différents. Pour prendre en compte cette hétérogénéité, la méthode la plus simple consiste à supposer que la surface est composée par une distribution de différents types de sites caractérisés par leur énergie d'adsorption. L'hypothèse majeure est que chaque type de site se remplit indépendamment des autres. Par conséquent, on peut définir pour chaque type de site un isotherme d'adsorption en fonction de la pression d'adsorbat ou de sa concentration, et l'isotherme d'adsorption total est une somme pondérée de l'adsorption dans tous les types de site [3] - [28] :

dans laquelle est l'isotherme d'adsorption dans les sites ayant une énergie d'interaction , et est la fonction de distribution des sites, c'est-à-dire la fraction de sites ayant une énergie d'interaction . L'isotherme local peut être l'une des nombreuses autres équations qui ont été proposées pour l'adsorption. Dans le cas des équations simples (Freundlich, Tóth, etc), il existe des solutions analytiques si on suppose que et que [29].





Pour la détermination de la distribution en taille des mésopores, la méthode la plus utilisée est celle de Barret, Joyner et Halenda[27]. Ces chercheurs ont supposé que l'on a des pores cylindriques dans lesquels l'adsorption se fait par condensation capillaire, on peut donc appliquer l'équation de Kelvin reliant le rayon des pores à la pression d'équilibre de l'adsorbat. Cette méthode, appelée BJH par référence aux auteurs, est la plus utilisée pour l'étude de la distribution en taille des pores ayant une taille supérieure à 4 nm (puisque l'équation de Kelvin n'est plus juste en dessous de cette taille)[3].
Depuis le début du XXIe siècle, l'isotherme local est souvent calculé par des méthodes telles que la théorie de la fonctionnelle de densité[30] ou la méthode Monte-Carlo. Les méthodes mathématiques permettant de recalculer la distribution à partir de l'isotherme total expérimental ont été développées[31], ce qui permet de calculer la distribution en taille des pores à partir de la mesure de l'isotherme d'adsorption.
Adsorbants
Généralités
Un adsorbant est un matériau qui a été optimisé pour un certain type d'adsorption. Il n'existe donc pas une unique liste de caractéristiques permettant de définir un adsorbant, mais plutôt un très grand nombre de types de matériaux pouvant avoir des propriétés très différentes. Les propriétés que l'on peut définir sont:
- la capacité d'adsorption qui peut être liée à la surface spécifique ou au volume poreux ;
- l'énergie d'interaction qui conditionne le caractère plus ou moins réversible de l'adsorption, une énergie trop importante peut défavoriser une régénération de l'adsorbant par désorption ;
- la cinétique d'adsorption peut être un paramètre essentiel dans certains procédés tels que la séparation de gaz par inversion de pression ;
- la tenue à la pression, à la température, la résistance à l'attrition peuvent être des facteurs essentiels pour la durée de vie de l'adsorbant ;
- la morphologie de l'adsorbant est un point essentiel si on utilise un lit d'adsorbant solide traversé par un fluide ;
- le coût financier et environnemental, la disponibilité locale de l'adsorbant doivent être pris en compte.
À l'heure actuelle, on trouve deux grandes classes de matériaux à grande surface utilisés comme adsorbant à grande échelle : les carbones activés (aussi nommés charbons actifs) et les oxydes tels que les zéolithes. Il existe de nombreux autres adsorbants basés sur des matériaux carbonés, sur des oxydes ou sur des polymères.
Les adsorbants sont généralement utilisés sous forme de granulés sphériques ou de tiges. Ils doivent avoir une bonne résistance à l'abrasion et à la température et avoir des pores de faibles diamètres, ce qui résulte en une surface spécifique élevée. Les adsorbants industriels les plus connus peuvent être classés en trois familles :
| Classe | Exemples | Propriétés |
|---|---|---|
| Adsorbants carbonés | Charbon actif et graphite | Hydrophobes et apolaires |
| Adsorbants oxygénés | Alumine activée, gel de silice et zéolithes | Hydrophiles et polaires |
| Adsorbants polymères | Souvent des polymères styréniques réticulés | Fonctions polaires et apolaires dans une matrice polymère |
Carbones activés
Un charbon actif est préparé en deux phases : la carbonisation d'un précurseur tel que le bois pour fabriquer un carbone, et l'activation de ce carbone par attaque chimique pour développer ses propriétés adsorbantes. Ces deux phases peuvent être réalisées successivement ou en même temps suivant les procédés de fabrication. En fonction du précurseur utilisé et des conditions de fabrication, on peut obtenir des charbons actifs ayant des propriétés différentes : surface spécifique, taille de pore, volume poreux, etc.
De façon générale, le charbon actif est un excellent adsorbant : sa capacité d’adsorption des molécules organiques et des gaz est remarquable, d’où son utilisation dans les masques de protection, dans l’antidote universel des Égyptiens ou dans des médicaments contre la dyspepsie. La plus grande partie de la production actuelle est utilisée pour la capture des polluants en solution aqueuse pour obtenir de l'eau potable, ou pour la purification de flux gazeux (épuration des fumées ou purification d'air).
Adsorbants oxygénés
Grâce à leur structure cristalline en feuillets, les argiles et les zéolites sont de bons adsorbants naturels.
Problèmes posés par l'adsorption et solutions
L’adsorption peut aussi être une propriété indésirable, un composé à faible concentration pouvant être complètement adsorbé sur les parois du récipient, de telle sorte qu’aux hautes dilutions, on ne retrouve plus trace de l’analyte. Ainsi, afin d’obtenir des résultats justes, les analystes utilisent des matériaux aussi inertes que possible, comme le polytétrafluoroéthylène (PTFE) (connu sous divers noms commerciaux comme « Teflon ») ou traitent préalablement les récipients, par exemple par silanisation du verre (inactivation de la silice avec du diméthyldichlorosilane).
Applications
Procédé de séparation
La physisorption sur les solides est utilisée pour la séparation et la purification des gaz et pour la séparation des solutés dans des liquides[32]. Dans le cas des gaz, le procédé de séparation par adsorption est un processus cyclique au cours duquel ont lieu alternativement l’adsorption d’un gaz par un solide ou un liquide à une pression et une température données, puis sa désorption. Selon la méthode de désorption utilisée, le procédé de séparation peut être[33] :
- adsorption à pression modulée (APM) (pressure swing adsorption, PSA, en anglais) : la désorption a lieu à une pression plus faible ;
- adsorption à température modulée (ATM) (temperature swing adsorption, TSA, en anglais) : la désorption a lieu à une température plus élevée.
- adsorption en phase liquide suivie d'une élution par modification du pH et/ou de la force ionique
- adsorption en phase liquide sur un charbon actif suivie d'une régénération externe dans un four (par exemple, la décoloration des sirops de glucose)
Autres applications
Parmi les autres applications pratiques faisant appel à l’adsorption, on peut citer :
- la catalyse hétérogène : le phénomène d’adsorption constitue la première étape des réactions nécessitant l’emploi d’un catalyseur solide. Ce phénomène peut alors jouer un rôle prédominant dans la cinétique de la réaction chimique ;
- la chromatographie d'adsorption ;
- la mesure de la surface spécifique des solides poreux et des poudres ;
- la stabilisation des colloïdes ;
- l’adhésion ;
- le stockage de chaleur (via l'adsorption par zéolithes).
Notes et références
- (en) Union internationale de chimie pure et appliquée (IUPAC), « Compendium of Chemical Terminology (Gold Book) : Adsorption », sur goldbook.iupac.org (consulté le ).
- (en) Union internationale de chimie pure et appliquée (IUPAC), « Compendium of Chemical Terminology (Gold Book) : Absorption », sur goldbook.iupac.org (consulté le ).
- (en) F. Rouquerol, J. Rouquerol, K. S. W. Sing et coll, "Adsorption by powders and porous solids: principles, methodology and applications", Academic Press 2nd edition, 2014.
- L-M Sun, F. Meunier, N. Brodu, M-H Manero, « Adsorption - Aspects théoriques », Techniques de l'ingénieur, J2730 V2, 2016.
- P. Le Cloarec, « Adsorption en traitement de l'air », Techniques de l'ingénieur, G1770 V1, 2003.
- (en) J. Rouquerol, D. Avnir, C. W. Fairbridge, D. H. Everett, et al., « Recommendations for the characterization of porous solids », Pure and Applied Chemistry, volume 66, numéro 8, 1994, p. 1739-1758.
- (de) Kayser, Heinrich, Über die Verdichtung von Gasen an Oberflächen in ihrer Abhängigkeit von Druck und Temperatur, Annalen der Physik und Chemie, volume 248, numéro 4, 1881, p. 526–537. doi:10.1002/andp.18812480404.
- C. W. Scheele "Chemische Adhandlung von der luft und dem feuer (1777).
- T. Lowitz Crell's Chemische Annalen vol 2, page 36 (1788).
- N. T. de Saussure "Beobachtungen über die absorption der gasarten durch verschiedene körper" Gilbert's Annalen der Physik, vol 47, pages 113-183.
- Z. A. Dabrowski "Adsorption - from theory to practice" Advances in Colloids and Interface Science, vol 93, pages 135-224 (2001).
- S. Brunauer, P. H. Emmet, E. Teller "Adsorption of gases in multimolecular layers", Journal of the American Chemical Society, vol 60(2), pages 309-319 (1938).
- N. R. Laine, F. J. Vastola, P. L. Walker "The importance of active surface area in the carbon oxygen reaction" Journal of Physical Chemistry, vol 67, pages 2030-2034 (1963).
- G. Crini "Non-conventional low-cost adsorbents for dye removal - a review" BioresourceTechnology, vol 97(9), pages 1061-1085.
- Z. A. Dabrowski, P. Podkoscielny, Z. Hubicki, M. .Barczak "Adsorption of phenolic compounds by activated carbon -a critical review" Chemosphere, vol 58, pages 1049-1070.
- S. Brunauer, L. S. Deming, W. S. Deming, E. Teller "on a theory of the van der Waals adsorption of gases" Journal of the American Chemical Society, vol 62, page 1723-1732 (1940).
- C. H. Giles, D. Smith, A. Huitson "A general treatment and classification of the solute adsorption isotherms. I. Theoretical" Journal of Colloid and Interface Science, vol 47(3), pages 755-765 (1974).
- C. H. Giles, T. H. MacEwan, S. N. Nakhwa, D. Smith "Studies in adsorption. Part XI. A system of classification of solution adsorption isotherms, and its use in diagnosis of adsorption mechanisms and in measurement of specific surface area of solids" journal of the Chemical Society, pages 3973-3993 (1960).
- K. Saleh, P. Guigon "Caractérisation et analyse des poudres - Propriétés physiques des solides divisés" Techniques de l'Ingénieur J2251 V1 (2009).
- L.-M. Sun, F. Meunier, N. Brodu, M.-H. Manero "Adsorption - Aspects théoriques" Techniques de l'Ingénieur, J2730 V2 (2016).
- Par exemple: Micromeritics , Quantachrome , MicrotracBel , etc.
- J. Pikunic, C. Clinard, N. Cohaut, K. E. Gubbins, et coll. "Structural modeling of porous carbons: constrained reverse Monte Carlo method" Langmuir, vol 19, pages 8565-8582.
- H. Freundlich "Kapillarchemie, eine Darstellung der Chemie der Kolloide und verwandter Gebiete" Akademische Verlagsgesellschaft (1909).
- I. Langmuir "The adsorption of gases on plane surfaces of glass, mica and platinum" Journal of the American Chemical Society, vol 40(9), pages 1361-1403 (1918).
- R. Sips "On the structure of a catalyst surface" Journal of Chemical Physics, vol 16(5), pages 490-495 (1948).
- K. Kaneko, C. Ishii, M. Ruike, H. Kubawara "Origin of superhigh surfacce area and microcrystalline graphitic structure of activated carbons" Carbon vol 30(7), pages 1075-1088 (1992).
- E. P. Barrett, L. G. Joyner, P. H. Halenda "The determination of pore volume and area distributions in porous substrances. 1. Computations from nitrogen isotherms" Journal of the American Chemical Society, vol 73(1), pages 373-380 (1951).
- M. Jaroniec "Adsorption on heterogeneous surfaces: the exponential equation for the overall adsorption isotherm" Surface Science, vol 50(2), pages 553-564 (1975).
- D. N. Misra, "New adsorption isotherms for heterogeneous surfaces" Journal of Chemical Physics, vol 52, page 5499 (1970).
- J. Jagiello, M. Thommes "Comparison of DFT characterization methods based on N2, Ar, CO2 and H2 adsorption applied to carbons with various pore size distributions" Carbon, vol 42(7), pages 1227-1232 (2004).
- J. Jagiello "Stable numerical solution of the adsorption integral equation" Langmuir, vol 10(8), pages 2778-2785 (1994).
- Xavier DUVAL, « ADSORPTION », Encyclopædia Universalis [en ligne], consulté le 21 février 2016. URL : http://www.universalis.fr/encyclopedie/adsorption/.
- Jimmy L. Humphrey, George E. Keller, Procédés de séparation, Techniques, sélection, dimensionnement, Collection: Technique et Ingénierie, Dunod/Industries et Technologies, 2001.