Synthèse totale de l'oseltamivir
La synthèse totale de l'oseltamivir correspond à la synthèse totale du principe actif antigrippal commercialisé par Hoffmann-La Roche sous le nom de marque « Tamiflu ». Sa production commerciale commence à partir de l'acide shikimique, une biomolécule issue de l'anis étoilé de Chine (fruit de Illicium verum) dont l'offre mondiale est limitée. À cause de cette offre limitée, des recherches sur d'autres voies de synthèse évitant l'acide shikimique sont en cours et en 2010, plusieurs de ces synthèses alternatives ont été publiées. Le contrôle de la stéréochimie est important : l'oseltamivir possède trois stéréocentres et l'isomère recherché est seulement l'un des 8 stéréoisomères possibles.
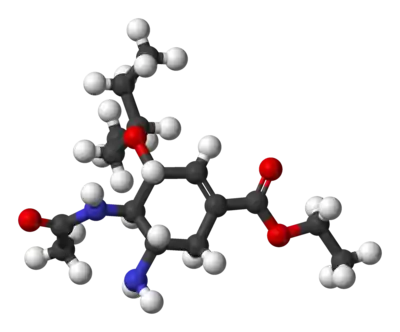
Production commerciale
La méthode courante de production de l'oseltamivir est basée sur les recherches de Gilead Sciences à partir de l'acide quinique naturel[1] - [2] et celles d'Hoffmann-La Roche à partir de l'acide shikimique[3].
Synthèse de Karpf-Trussardi
La méthode de production actuelle comprend deux étapes avec des azotures potentiellement dangereux car explosifs. Une synthèse sans azoture du Tamiflu Hoffmann-La Roche, publiée est décrite graphiquement ci-dessous[4]:
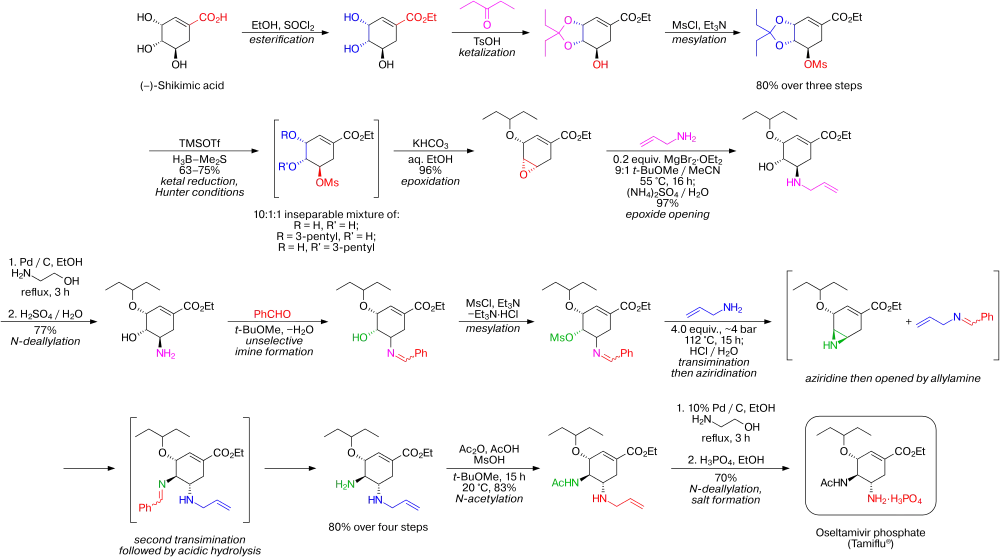
La synthèse commence à partir de l'acide (–)-shikimique. Le 3,4-pentylidène acétal mésylate, est préparé en trois étapes avec un rendement global de 80 % : estérification avec l'éthanol et le chlorure de thionyle, cétalisation avec l'acide paratoluènesulfonique (APTS) et la pentan-3-one, mésylation avec la triéthylamine et le chlorure de méthanesulfonyle. L'ouverture réductrice du cétal sous conditions de Hunter modifiées[5] produit un mélange inséparable de, majoritairement (> 80 %), mésylate diol, inintéressant et des mésylates isomériques monopentyl éther dont celui recherché ne correspond qu'à environ 8 % mol. du mélange total. L'époxyde correspondant est formé sous conditions basiques avec du bicarbonate de potassium. En utilisant le diéthyl étherate de bromure de magnésium, un acide de Lewis très bon marché (et communément préparé frais par addition de copeaux de magnésium métallique sur le 1,2-dibromoéthane dans un mélange benzène:diéthyl éther), l'époxyde est ouvert avec l'allylamine pour former l'aminoalcool-1,2 correspondant. Les solvants méthyl tert-butyl éther et acétonitrile, non miscibles à l'eau, sont utilisés pour simplifier la purification qui consiste à agiter le mélange réactionnel avec une solution aqueuse 1M de sulfate d'ammonium. Une réduction sur palladium, catalysée avec de l'éthanolamine suivie par une purification acide fournit l'aminoalcool-1,2 déprotégé (N-désallylation). Cet aminoalcool est converti directement en l'allyl-diamine correspondante par une intéressante séquence en cascade qui commence par une imination (formation d'une imine) non sélective du benzaldéhyde avec élimination de l'eau par l'azéotrope qu'elle forme avec le méthyl tert-butyl éther. Puis une mésylation suivie de l'élimination du chlorhydrate de triéthylamine, un coproduit solide, forme un intermédiaire qui subit une aziridination (formation d'un cycle aziridine) simultanément à une transimination avec un autre équivalent d'allylamine. Avec l'acide méthylsulfonique libéré, l'aziridine s'ouvre proprement pour former la diamine qui subit immédiatement une seconde transimination. Une hydrolyse acide élimine l'imine. Une acylation sélective avec l'anhydride acétique (sous conditions tamponnées, le groupe 5-amino est protoné en raison de son pKa de 4,2 -considérablement plus bas qu'un pKa attendu aux alentours de 8 pour ce genre de groupe- ce qui empêche son acétylation) donne le produit N-acétylé désiré sous forme cristalline après purification. Finalement, une désallynation comme précédemment, produit l'oseltamivir free base qui est converti en le phosphate d'oseltamivir désiré par traitement avec l'acide phosphorique. Ce produit final est obtenu avec une très bonne pureté de 99,7 % et avec un rendement général de 17 à 22 % à partir de l'acide (–)-shikimique.
Il est à noter que cette synthèse permet d'éviter l'utilisation de réactifs et d'intermédiaires azotures potentiellement explosifs mais la synthèse effectivement mise en œuvre par Hoffmann-La Roche les utilise. Hoffmann-La Roche connaît d'autres voies vers l'oseltamivir qui n'impliquent pas l'utilisation d'acide shikimique comme composé de départ possédant une chiralité définie. Une de ces voies exploite une réaction de Diels-Alder impliquant l'acrylate d'éthyle et des furanes, une autre voie l'acide isophtalique qui implique une hydrogénation catalytique et une désymétrisation enzymatique.
Synthèse de Corey
En 2006, l'équipe d'Elias James Corey publie une nouvelle route contournant l'acide shikimique et partant du butadiène et de l'acide acrylique[6]. Les inventeurs ont choisi de ne pas breveter cette procédure en 11 étapes, décrite ci-dessous :
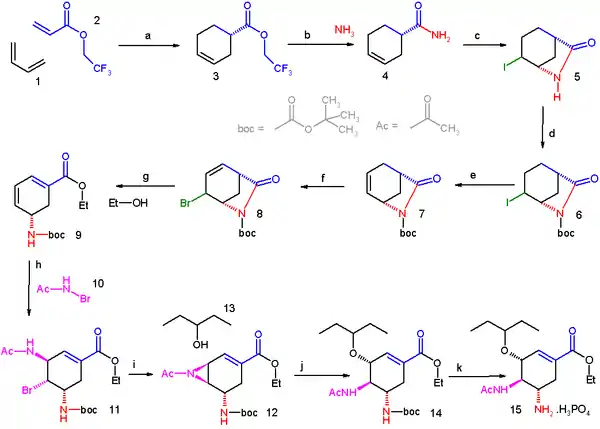
Le butadiène (1) réagit dans une réaction de Diels-Alder asymétrique avec l'ester acrylate du 2,2,2-trifluoroéthanol (2) et catalysé par le catalyseur CBS. L'ester (3) est converti en l'amide (4) par réaction avec l'ammoniac et l'étape suivante vers le lactame (5) est une iodolactamisation initiée par le triflate de triméthylsilyle. Le groupe protecteur BOC est ajouté sur le groupe amide par réaction avec l'anhydride de BOC dans (6) et le substituant iode est éliminé par une réaction d'élimination avec DBU pour former l'alcène (7). Un atome de brome est introduit dans (8) par une bromation allylique avec le NBS et le groupe amide est coupé avec de l'éthanol et du carbonate de césium, accompagné de l'élimination du bromure pour former l'ester éthylique de diène (9). La nouvelle double liaison formée est fonctionnalisée par le N-bromoacétamide (10) dans une réaction catalysée avec du bromure d'étain(IV) qui permet un contrôle complet de la stéréochimie de (11). Dans l'étape suivante, l'atome de brome est remplacé par l'atome d'azote du substituant acétamide avec la base forte KHMDS ce qui fournit l'aziridine (12) qui, à son tour, est ouverte par réaction avec le pentan-3-ol (13) vers l'éther (14). Dans l'étape finale, le groupe BOC est retiré par l'acide phosphorique et le phosphate d'oseltamivir (15) est formé.
Synthèse de Shibasaki
En 2006 aussi, l'équipe de Masakatsu Shibasaki de l'université de Tokyo publie une synthèse qui évite aussi l'acide shikimique mais emploie des azotures[7] - [8].
 |
 | |
| Synthèse de Shibasaki du Tamiflu Part. I | Part. II | |
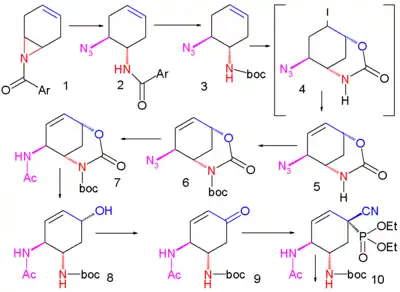
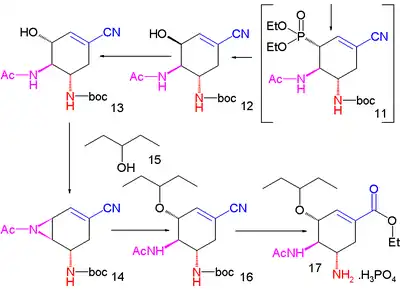
Une méthode améliorée publiée en 2007 commence par la désymétrisation énantiosélective de l'aziridine (1) avec l'azoture de triméthylsilyle (TMS–N3) et un catalyseur chiral pour former l'azoture (2). Le groupe amide est protégé par un groupe BOC introduit par l'anhydride de BOC et la DMAP dans (3) dont la iodolactamisation avec de l'iode et du carbonate de potassium donne initialement l'intermédiaire instable (4) qui se réarrange en le carbamate cyclique (5) après élimination d'iodure d'hydrogène (HI) avec le DBU.
Le groupe amide est reprotégé avec BOC dans (6) et le groupe azoture est converti en acétamide dans (7) par acylation réductrice avec l'acide thioacétique et la 2,6-lutidine. Le carbonate de césium accomplit l'hydrolyse du groupe carbamate pour former l'alcool (8) qui est ensuite oxydé en la cétone (9) par le periodinane de Dess-Martin. Une cyanophosphorylation avec le cyanophosphate de diéthyle (numéro CAS ) modifie le groupe cétone en le cyanophosphate (10) ouvrant la voie à un réarrangement allylique intramoléculaire vers le phosphate β-allylique instable (11) (dans le toluène en tube scellé) qui est hydrolysé en l'alcool (12) par le chlorure d'ammonium. Ce groupe hydroxyle a une stéréochimie erronée et est donc inversé dans une réaction de Mitsunobu avec l'acide 4-nitrobenzoïque (numéro CAS ) suivie par l'hydrolyse du para-nitrobenzoate pour former (13).
Une seconde réaction de Mitsunobu forme alors l'aziridine (14), prête pour une réaction d'ouverture de cycle avec le pentan-3-ol (15) catalysée par du trifluorure de bore et qui forme l'éther (16). Dans l'étape finale, le groupe protecteur BOC est retiré (HCl) puis par réaction avec l'acide phosphorique, le phosphate d'oseltamivir (17) est formé.
Synthèse de Fukuyama
Une approche publiée en 2007[9] comme celle de Corey commence par une réaction de Diels-Alder asymétrique cette fois avec la pyridine et l'acroléine comme réactifs de départ.
 |
 | |
| Synthèse de Fukuyama du Tamiflu Part. I | Part. II | |
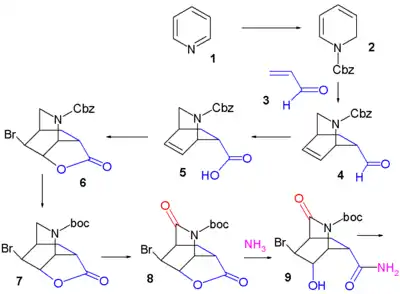
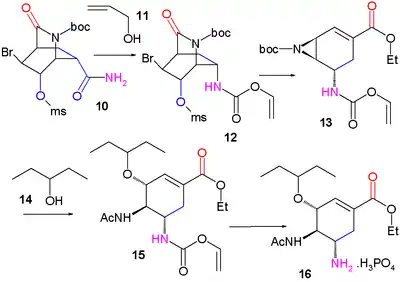
La pyridine (1) est réduite avec le borohydrure de sodium (NaBH4) en présence de chloroformiate de benzyle pour former la dihydropyridine (2) protégée par un groupe Cbz. La réaction de Diels-Alder asymétrique avec l'acroléine (3) est menée en présence de catalyseur de McMillan pour former strictement l'isomère endo de l'aldéhyde (4) qui est oxydé en l'acide carboxylique (5) avec du chlorite de sodium (NaClO2), du dihydrogénophosphate de potassium (KH2PO4) et 2-méthylbut-2-ène ((CH3)2C=CHCH3). L'addition de brome donne par halolactamisation la produit (6) et après replacement du groupe protecteur Cbz par BOC le produit (7) (hydrogénolyse en présence de dicarbonate de di-tert-butyle), un groupe carbonyle est introduit dans l'intermédiaire (8) avec de l'oxyde de ruthénium(IV) catalytique et du periodate de sodium (NaIO4) comme catalyseur sacrificiel. L'addition d'ammoniac ouvre le groupe lactame et forme l'amide (9) dont le groupe hydroxyle est mésylaté (méthanesulfonaté) dans (10).
Dans l'étape suivante, du diacétate d'iodobenzène (numéro CAS ) est ajouté et convertit l'amide par un réarrangement d'Hofmann en le carbamate d'allyle (12) après capture de l'intermédiaire isocyanate avec l'alcool allylique (11). À l'ajout d'éthanolate de sodium dans l'éthanol, trois réactions se déroulent simultanément : coupure de l'amide protégé par BOC qui forme un nouveau groupe ester d'éthyle, déplacement du groupe mésylate par la nouvelle amine protégée par BOC vers un groupe aziridine et réaction d'élimination qui forme la double liaison alcénique de (13) avec libération de HBr. Dans les deux dernières étapes, le cycle aziridine est ouvert par le pentan-3-ol (14) et le trifluorure de bore pour former l'aminoéther (15) avec le groupe BOC remplacé par un groupe acétyle et après retrait du groupe protecteur de l'autre amine (Pd/C, Ph3P et acide 1,3-diméthylbarbiturique dans l'éthanol) et addition d'acide phosphorique, le Tamiflu (16) est obtenu.
Synthèse de Trost
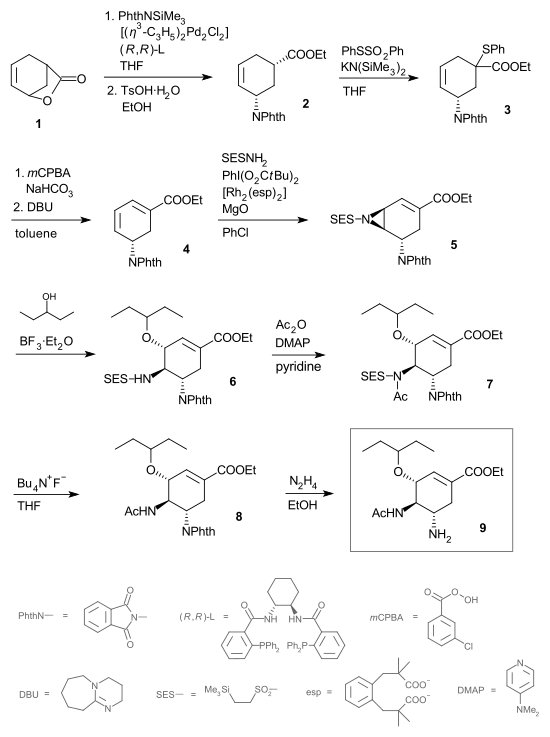
En 2008, le groupe de Barry M. Trost de l'Université Stanford publie la plus courte voie de synthèse de l' oseltamivir de bonne stéréoisomérie (9) à ce jour[10].
Synthèse de Hayashi
En 2009, Hayashi Yujiro et son équipe mettent au point avec succès une voie de synthèse efficace et peu coûteuse pour préparer l'oseltamivir de bonne stéréoisomérie (3R,4R,5S). Leur objectif était de concevoir une procédure adaptée à la production à grande échelle. En gardant à l'esprit le coût, le rendement et le nombre d'étapes de synthèse, une synthèse totale énantiosélective du l'oseltamivir a été réalisée grâce à trois réactions one-pot[11] - [12]. Ces réactions monotopes leur a donc permis d'effectuer plusieurs étapes de réaction en même temps, ce qui a finalement minimisé le nombre d'étapes de purification nécessaires et les pertes qu'elles engendrent et permis un fort gain de temps.
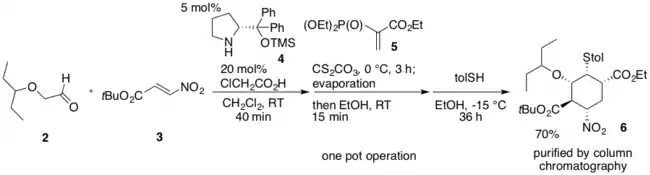
Dans la première synthèse one-pot, l'équipe de Hayashi Yujiro commence par utiliser le diphénylprolinol silyl éther (4)[13] comme catalyseur organique pour effectuer une addition de Michael asymétrique de l'alcoxyaldéhyde (2) et du nitroalcène (3) et donnant l'adduit de Michael de bonne énantiomérie. Lors de l'addition d'un dérivé de phosphate de diéthyle et de vinyle (5) à l'adduit de Michael, une réaction de Michael en domino et une réaction de Horner-Wadsworth-Emmons se produisent en raison du dérivé avec le groupe phosphonate issu de 5. Pour transformer des sous-produits indésirables en le dérivé cyclohexéncarboxylate d'éthyle désiré, le mélange du produit et des sous-produits est traité avec du carbonate de césium, Cs2CO3 dans de l'éthanol. Cela provoque une rétro-réaction Michael sur un des sous-produits et une rétro-réaction aldol accompagnée d'une réaction de Horner-Wadsworth-Emmons sur un autre. Les deux sous-produits sont ainsi convertis avec succès en le dérivé souhaité. Finalement, l'addition de p-toluènethiol en présence du Cs2CO3 donne 6 avec un rendement de 70% après avoir été purifié par chromatographie sur colonne, avec un bon excès énantiomérique pour le stéréoisomère souhaité[11].
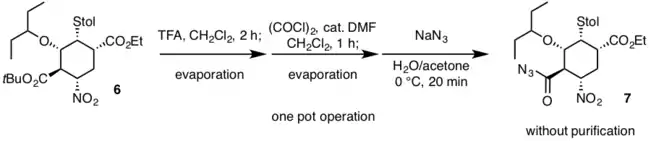
Dans la deuxième opération monotope, l'acide trifluoroacétique est utilisé en premier lieu pour déprotéger l'ester tert-butylique de 6 puis tout excès de réactif est éliminé par évaporation. L'acide carboxylique produit à la suite de cette déprotection est ensuite converti en un chlorure d'acyle par le chlorure d'oxalyle et une quantité catalytique de diméthylformamide, DMF. Enfin, l'addition d'azoture de sodium produit l'acylazoture 7 sans aucune purification[11].
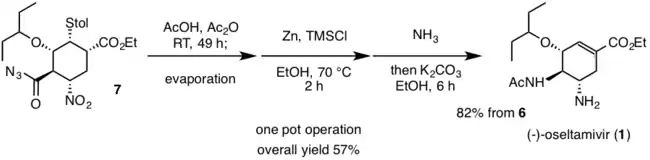
La synthèse monotope finale commence par un réarrangement de Curtius de l'acylazoture (7) pour produire un groupe fonctionnel isocyanate à température ambiante. Le dérivé isocyanate réagit alors avec l'acide acétique pour donner le fragment acétylamino souhaité trouvé dans l'oseltamivir (1). Ce réarrangement de Curtius en domino et la formation de cette amide se produisent en l'absence de chaleur, ce qui est extrêmement bénéfique pour réduire tout risque possible d'accident dû à la chaleur. Le groupe nitro de 7 est réduit en l'amine souhaitée dans 1 avec le couple zinc et chlorure d'hydrogène, HCl. En raison des conditions dures de la réduction nitro, l'ammoniac, NH3 est ajoutée pour réduire la réaction. Du carbonate de potassium est ensuite ajouté pour donner 1, via une rétro-réaction de Michael du thiol. L'oseltamivir est ensuite purifié par une extraction acide / base. Le rendement global pour la synthèse totale de l'oseltamivir est de 57%[11]. L'utilisation de réactifs bon marché et non dangereux a permis d'obtenir une voie de synthèse efficace et à haut rendement permettant de produire une grande quantité de nouveaux dérivés dans l'espoir de lutter contre les virus résistants à l'oseltamivir.
Synthèse de Kongkathip
Cette étude publiée en 2015, un nouveau procédé de fabrication, plus rapide et plus efficient, a été mis au point, à partir de la molécule D-glucose, produit très abondant et très peu coûteux[14].
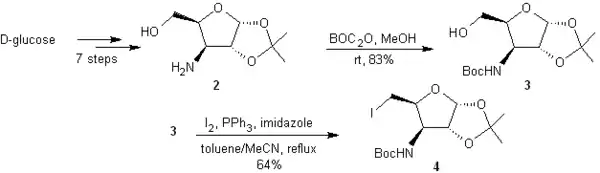
En 7 étapes à partir du D-glucose, un étude précédente permet d'obtenir le 3-amino-3-désoxy-1,2-O-(1-méthyléthylidène)- α-D-xylofuranose (2)[15] dont la fonction amine est protégée par BOC via une réaction avec BOC2O pour pouvoir le ioder et obtenir 4.

4 subit une réaction en deux étapes, dérivée de Bernet-Vasella qui, elle, fournit un mélange de 5a et 5b équimolaire. Le zinc activé permet la fragmentation réductrice de 4, la poudre d'indium promeut le couplage entre l'aldéhyde intermédiaire et le bromoacrylate d'éthyle et finalement un mélange 3:1 en faveur du bon stéréoisomère est obtenu avec un rendement global de 82%. 5a et 5b sont séparés par chromatographie flash sur colonne.
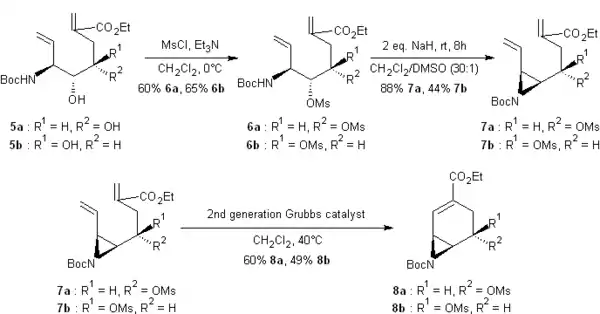
La protection des groupes hydroxyle de 5a et 5b par traitement avec le chlorure de méthanesulfonyle, MsCl fournit les mésylates 6a et 6b avec un bon rendement. Ces derniers sont traités avec 2 équivalents d'hydrure de sodium, NaH dans un mélange 30:1 de dichlorométhane et de diméthylsulfoxyde, DMSO à température ambiante et les aziridines 7a et 7b désirées sont obtenues. Celles-ci mises à réagir avec un catalyseur de Hoveyda-Grubbs de seconde génération et contenant des ions ruthénium, subissent une métathèse qui les cyclisent, fournissant ainsi les bicycles 8a et 8b avec des rendements de 60% et 49%, respectivement.
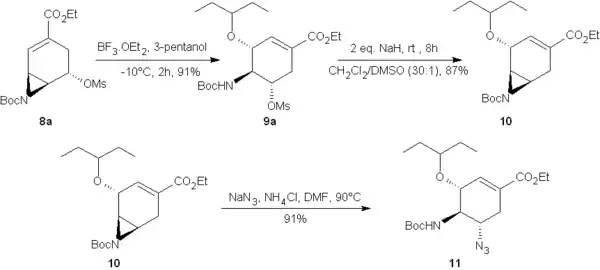
Les N-BOC aziridines 8a et 8b sont immédiatement mises en présence de BF3.OEt2 et d'un large excès de pentan-3-ol qui provoquent l'ouverture régio- et stéréospécifique du cycle aziridine avec l'insertion correcte du pentan-3-ol dans 9a et 9b.
9a réagit en deux étapes pour fournir le dérivé azoture (11) tandis que 9b le fournit directement (ci-dessus)
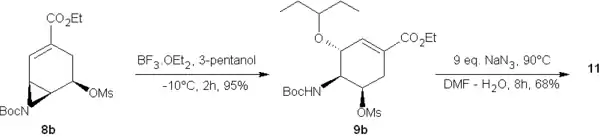
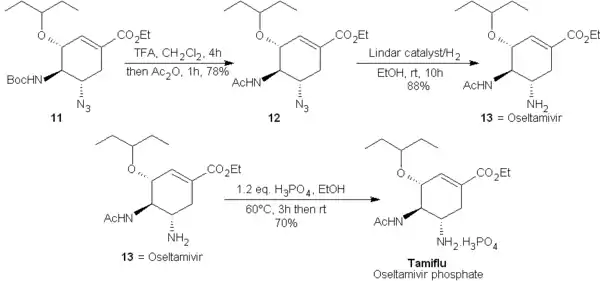
Finalement, l'amine-BOC de 11 est converti en l'acétyl-amine présente sur l'oseltamivir par traitement avec l'acide trifluoroacétique, TFA dans le dichlorométhane puis acétylé dans 12 avec de l'anhydride acétique. Le groupe azoture est ensuite réduit par hydrogénation avec un catalyseur de Lindlar et fournit alors l'oseltamivir (13) qui peut être facilement converti en le phosphate d'oseltamivir, Tamiflu par traitement avec 1,2 équivalent d'acide phosphorique dans l'éthanol. Le rendement global des 9 étapes pour passer de 4 au Tamiflu est de 7,2%[14].
Notes et références
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Oseltamivir total synthesis » (voir la liste des auteurs).
- Kim, C. U.; Lew, W.; Williams, M. A.; Liu, H.; Zhang, L.; Swaminathan, S.; Bischofberger, N.; Chen, M. S.; Mendel, D. B.; Tai, C. Y.; Laver, W. G.; Stevens, R. C.; Influenza Neuraminidase Inhibitors Possessing a Novel Hydrophobic Interaction in the Enzyme Active Site: Design, Synthesis, and Structural Analysis of Carbocyclic Sialic Acid Analogues with Potent Anti-Influenza Activity, J. Am. Chem. Soc., 1997, vol. 119(4), pp. 681-690. DOI 10.1021/ja963036t
- John C. Rohloff, Kenneth M. Kent, Michael J. Postich, Mark W. Becker, Harlan H. Chapman, Daphne E. Kelly, Willard Lew, Michael S. Louie, Lawrence R. McGee, Ernest J. Prisbe, Lisa M. Schultze, Richard H. Yu, and Lijun Zhang, Practical Total Synthesis of the Anti-Influenza Drug GS-4104, J. Org. Chem., 1998, vol. 63(13), pp. 4545-4550. DOI 10.1021/jo980330q.
- Abrecht, S.; Harrington, P.; Iding, H.; Karpf, M.; Trussardi, R.; Wirz, B.; Zutter, U.; Chimia, 2004,vol. 58, p. 621.
- Martin Karpf and René Trussardi, New, Azide-Free Transformation of Epoxides into 1,2-Diamino Compounds: Synthesis of the Anti-Influenza Neuraminidase Inhibitor Oseltamivir Phosphate (Tamiflu), J. Org. Chem., 2001, vol. 66(6), pp. 2044-2051. DOI 10.1021/jo005702l.
- (en) Birgit Bartels and Roger Hunter, « A selectivity study of activated ketal reduction with borane dimethyl sulfide », J. Org. Chem., vol. 58, , p. 6756 (DOI 10.1021/jo00076a041)
- Ying-Yeung Yeung, Sungwoo Hong, and E. J. Corey, A Short Enantioselective Pathway for the Synthesis of the Anti-Influenza Neuramidase Inhibitor Oseltamivir from 1,3-Butadiene and Acrylic Acid, J. Am. Chem. Soc., 2006, vol. 128(19), pp. 6310-6311. DOI 10.1021/ja0616433.
- Yuhei Fukuta, Tsuyoshi Mita, Nobuhisa Fukuda, Motomu Kanai, and Masakatsu Shibasaki, De Novo Synthesis of Tamiflu via a Catalytic Asymmetric Ring-Opening of meso-Aziridines with TMSN3, J. Am. Chem. Soc., 2006, vol. 128(19), pp. 6312-6313. DOI 10.1021/ja061696k.
- Tsuyoshi Mita, Nobuhisa Fukuda, Francesc X. Roca, Motomu Kanai, and Masakatsu Shibasaki, Second Generation Catalytic Asymmetric Synthesis of Tamiflu: Allylic Substitution Route, Org. Lett., 2007, vol. 9(2), pp. 259-262. DOI 10.1021/ol062663c.
- Nobuhiro Satoh, Takahiro Akiba, Satoshi Yokoshima, Tohru Fukuyama, A Practical Synthesis of (–)-Oseltamivir, Angew. Chem. Int. Ed., 2007, vol. 46, pp. 5734-5736. DOI 10.1002/anie.200701754.
- Barry M.Trost, Ting Zhang, A Concise Synthesis of (−)-Oseltamivir, Angew. Chem. Int. Ed., 2008, vol. 47, pp. 1-4.DOI 10.1002/anie.200800282.
- Hayato Shikawa, Takaki Suzuki, Yujiro Hayashi, High-Yielding Synthesis of the Anti-Influenza Neuramidase Inhibitor (−)-Oseltamivir by Three "One-Pot" Operations, Angewandte Chemie International Edition, 2009, vol. 48(7), pp. 1304–1307. DOI 10.1002/anie.200804883, (ISSN 1521-3773).
- Pedro Laborda, Su-Yan Wang, Josef Voglmeir, Influenza Neuraminidase Inhibitors: Synthetic Approaches, Derivatives and Biological Activity, Molecules, 2016, vol. 21(11), p. 1513. DOI 10.3390/molecules21111513.
- Yujiro Hayashi, Hiroaki Gotoh, Takaaki Hayashi, Mitsuru Shoji, Diphenylprolinol Silyl Ethers as Efficient Organocatalysts for the Asymmetric Michael Reaction of Aldehydes and Nitroalkenes, Angewandte Chemie International Edition, 2005, vol. 44(27), pp. 4212–4215. DOI 10.1002/anie.200500599, (ISSN 1521-3773).
- Boonsong Kongkathip, Sunisa Akkarasamiyo et Ngampong Kongkathip, A new and efficient asymmetric synthesis of oseltamivir phosphate (Tamiflu) from D-glucose, Tetrahedron, 2015, vol. 71(16), pp. 2393-2399. DOI 10.1016/j.tet.2015.02.081, résumé.
- Eva Raluy, Oscar Pàmies, Montserrat Diéguez, Modular Furanoside Phosphite‐Phosphoroamidites, a Readily Available Ligand Library for Asymmetric Palladium‐Catalyzed Allylic Substitution Reactions. Origin of Enantioselectivity, Adv. Synth. Catal., 2009, vol. 351(10), pp. 1648-1670. DOI 10.1002/adsc.200900073.