Hydrogénation
L'hydrogénation est une réaction chimique qui consiste en l'addition d'une molécule de dihydrogène (H2) à un autre composé. Cette réaction est habituellement employée pour réduire ou saturer des composés organiques. Elle nécessite en général une catalyse, les réactions sans catalyse nécessitant de très hautes températures.

On appelle la réaction inverse de l'hydrogénation la déshydrogénation. Les réactions où des liaisons sont brisées tandis que de l'hydrogène est additionné sont appelées hydrogénolyses (cette réaction pouvait s'appliquer aux liaisons carbone-carbone comme aux liaisons carbone-hétéroatome — O, N, X). L'hydrogénation diffère de la protonation ou de l'addition d'hydrure : dans l'hydrogénation, le produit ou les produits ont la même charge que les réactifs.
La réaction d'hydrogénation utilise généralement du dihydrogène gazeux comme source d'hydrogène. Cependant cette réaction étant relativement importante, de nombreuses techniques ont été développées et certaines d'entre elles utilisent d'autres sources que H2 ; on appelle alors ce type de réaction hydrogénation par transfert.
La réaction d'hydrogénation est utilisée dans de nombreux domaines, en particulier en pétrochimie (transformation d'alcènes en alcanes) et dans l'industrie agroalimentaire (hydrogénation de graisses insaturées en graisses saturées).
Historique
La réaction d'hydrogénation a été décrite pour la première fois en 1897 par le chimiste français Paul Sabatier qui est considéré comme le père des procédés d'hydrogénation. En 1897, il a découvert que l'introduction de traces de nickel comme catalyseur facilitait l'addition de dihydrogène sur des hydrocarbures gazeux, procédé connu à présent sous le nom de « procédé de Sabatier ». Ses nombreux travaux sur l'hydrogénation des composés organiques lui ont valu entre autres l'attribution du prix Nobel de chimie en 1912, conjointement avec un autre chimiste français, Victor Grignard.
L'hydrogénation a alors été utilisée dans de nombreux procédés :
- Wilhelm Normann a obtenu en 1902 en Allemagne, et en 1903 au Royaume-Uni un brevet sur l'hydrogénation des huiles liquides, ce qui allait devenir une nouvelle industrie à l'échelle mondiale ;
- le procédé Haber-Bosch, d'une grande importance commerciale, décrit pour la première fois en 1905, utilise l'hydrogénation du diazote ;
- dans le procédé Fischer-Tropsch, décrit en 1922, le monoxyde de carbone obtenu facilement à partir du charbon est hydrogéné en combustibles liquides.
En 1922, Voorhees et Adams ont décrit un appareil permettant d'effectuer des hydrogénations à pression élevée[2]. L'agitateur de Parr (Parr shaker), le premier appareil permettant l'hydrogénation à hautes température et pression, basé sur les travaux de Voorhees et Adams, a été commercialisé en 1926 et reste d'usage courant. En 1938, Otto Roelen a décrit le procédé oxo (hydroformylation) qui utilise à la fois l'addition d'hydrogène et de monoxyde de carbone sur les alcènes pour produire des aldéhydes. Puisque ce procédé implique la formation de liaison C-C, il est, ainsi que ses nombreuses déclinaisons (voir carbonylation), toujours d'une grande actualité[3]. Les années 1960 ont vu le développement des catalyseurs homogènes, avec par exemple le catalyseur de Wilkinson. Dans les années 1980, l'hydrogénation asymétrique de Noyori représente l'une des premières applications de l'hydrogénation en synthèse asymétrique, une branche en pleine croissance permettant la production de produits chimiques spécifiques (chimie fine). En 2004, le H-Cube, un appareil sophistiqué à hydrogénation, est développé.
Réaction
Les réactions d'hydrogénation sont en général très favorables thermodynamiquement. Par exemple, pour la réaction d'hydrogénation d'un alcène, l'éthylène (C2H4 + H2 → C2H6), si la baisse du facteur entropique (ΔrS°=−120,9 J·K-1·mol-1) défavorise la réaction, une forte exothermicité (ΔrH°=−136,95 kJ·mol-1) la compense largement pour obtenir à température ambiante une enthalpie libre de réaction très négative (ΔrG°=−100,92 kJ·mol-1). Cependant cette réaction, en raison de la grande stabilité de la molécule de dihydrogène (ΔH°diss (H2)=434 kJ·mol-1), nécessite soit une grande quantité d'énergie (haute température) soit une catalyse. En règle générale, la deuxième solution est presque toujours préférée, ainsi, la réaction démarre en présence de trois espèces : le réactif insaturé à hydrogéner, la source d'hydrogène (H2 le plus souvent) et le catalyseur.
Réactifs
Les composés susceptibles d'être hydrogénés sont les composés chimiques possédant une ou plusieurs liaisons multiples. Dans certains cas, en particulier pour les composés possédant une liaison triple, le produit final dépend également des conditions de l'hydrogénation. Par exemple, dans le cas des alcynes, si ceux-ci sont hydrogénés de façon douce, ils produiront des alcènes alors que dans des conditions plus rudes, ils produiront directement les alcanes correspondants.
| alcène, R2C=CR'2 | alcane, R2CHCHR'2 |
| alcyne, RCCR | alcène, cis-RHC=CHR' ou alcane, RH2CCR'H2 |
| aldéhyde, RCHO | alcool primaire, RCH2OH |
| cétone, R2CO | alcool secondaire, R2CHOH |
| ester, RCO2R' | deux alcools, RCH2OH, R'OH |
| imine, RR'CNR" | amine, RR'CHNHR" |
| amide, RC(O)NR'2 | amine, RCH2NR'2 |
| nitrile, RCN | imine, RHCNH |
| Nitro, RNO2 | amine, RNH2 |
Source en hydrogène
La source d'hydrogène la plus commune est évidemment le dihydrogène gazeux, H2, généralement conservé en cylindre pressurisé. La réaction d'hydrogénation nécessite d'ailleurs fréquemment une pression en dihydrogène supérieure à une atmosphère. Le dihydrogène gazeux est produit industriellement par reformage d'hydrocarbures[4].
L'hydrogène peut aussi, dans certaines applications spécialisées, être extrait (on parle de « transfert ») de « donneurs en hydrogène » qui sont souvent des solvants comme l'hydrazine, le dihydronaphtalène, le dihydroanthracène, l'isopropanol, l'acide méthanoïque[5], ou encore le cyclohexadiène. En synthèse organique, l'hydrogénation par transfert est utile pour la réduction de composés insaturés polaires comme les cétones, les aldéhydes ou les imines.
Catalyseurs


Si la réaction est favorable thermodynamiquement, elle est difficile à démarrer du fait de la grande stabilité du dihydrogène. Ainsi, sauf exception, aucune réaction ne se produit entre le dihydrogène H2 et le composé organique à une température inférieure à 480 °C et en l'absence de catalyseur. D'une part chauffer le milieu réactionnel à haute température est peu pratique (quoique faisable), et d'autre part certains composés organiques ne peuvent supporter une température élevée. La solution privilégiée consiste donc à utiliser un catalyseur. Ce catalyseur va se lier à la fois au dihydrogène et au composé insaturé, et ainsi les « aider » à réagir ensemble.
Les métaux du groupe du platine, en particulier le platine, le palladium, le rhodium et le ruthénium sont des catalyseurs particulièrement actifs, et qui agissent à basse température et faible pression en H2.
Des catalyseurs à partir de métaux non précieux ont été développés, en particulier à base de nickel (comme le nickel de Raney ou le nickel d'Urushibara) comme alternative plus économique, mais ceux-ci sont généralement plus lents ou requièrent des températures plus hautes et une pression en dihydrogène plus importante (en particulier le nickel de Raney[6] - [7]). Un compromis est donc nécessaire entre la réactivité du catalyseur (donc la vitesse de réaction) et le prix de celui-ci, mais aussi le prix de l'équipement pour travailler à haute pression. Il existe aussi des catalyseurs à base de cobalt, de fer et de chromite de cuivre ou de zinc.
Les catalyseurs sont classés en deux grandes familles :
- les catalyseurs qui se dissolvent dans le solvant où se trouve le composé insaturé à hydrogéner ; on parle alors de catalyseur homogène ;
- les catalyseurs qui restent des solides en suspension dans le solvant qui contient le composé insaturé à hydrogéner, ou les catalyseurs (solides) utilisés pour des réactions en phase gazeuse (réactifs gazeux) ; on parle alors de catalyseur hétérogène.
Poison de catalyseur
On appelle poison de catalyseur toute substance qui réagit avec un site actif de la surface d'un catalyseur, et qui bloque son action partiellement ou totalement. De telles substances peuvent être responsables de l'échec d'une hydrogénation catalytique, mais peuvent aussi servir à rendre le catalyseur moins réactif et plus sélectif. C'est par exemple le cas du catalyseur de Lindlar, un catalyseur hétérogène dont certains sites sont désactivés par l'action de l'acétate de plomb. Des poisons « classiques » sont les composés sulfurés (thiols par exemple, dont l'effet négatif sur le catalyseur augmente avec la longueur de la chaine carbonée). Basiquement, les poisons de catalyseurs peuvent être classés en trois groupes :
- les composés comportant des éléments non métalliques comme de l'azote, du phosphore, de l'arsenic, de l'antimoine, du soufre, du sélénium, du tellure, du brome, l'iode et parfois du chlore ;
- les composés et ions métalliques. Par exemple Cu2+, Cu+, Ag+, Au+, Zn2+, Cd2+, Hg22+, Hg2+, Mn2+, Fe2+, Co2+ et Ni2+ sont des poisons pour des catalyseurs au platine ;
- les composés complexant les métaux, par exemple les ions cyanure, le monoxyde de carbone ou le benzène.
Catalyseur homogène
Parmi les catalyseurs homogènes, on peut notamment citer le catalyseur de Wilkinson, à base de rhodium, ou encore le catalyseur de Crabtree à base d'iridium.
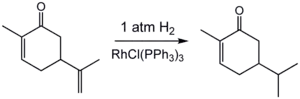 Hydrogénation de la carvone.
Hydrogénation de la carvone.
L'hydrogénation est sensible à l'encombrement stérique, ce qui explique l'absence de réaction avec la double liaison endocyclique.
L'activité et la sélectivité des catalyseurs homogènes est ajustée en faisant varier les ligands. Pour un composé prochiral, la sélectivité de la catalyse peut être ajustée de telle façon que la production d'un seul énantiomère soit favorisée. Les techniques utilisant des catalyseurs homogènes ont réellement commencé à se développer dans les années 1960, l'essentiel des catalyseurs utilisés étant alors des catalyseurs hétérogènes. Bien que les techniques se soient développées, les catalyseurs homogènes sont moins réactifs que les catalyseurs hétérogènes, et ces derniers sont donc toujours les plus employés.
Catalyseur hétérogène
Les catalyseurs hétérogènes sont les plus utilisés pour l'hydrogénation dans l'industrie. Comme pour les catalyseurs homogènes, leur activité peut être ajustée en changeant l'environnement autour du métal c'est-à-dire sa sphère de coordination. Ainsi, par exemple les différentes faces d'un catalyseur hétérogène cristallin peuvent avoir des activités distinctes. L'avantage évident est que l'on peut facilement se débarrasser du catalyseur par une simple filtration.
En chimie organique, on utilise très souvent le palladium sur charbon à 5 ou 10 % (abrégé « Pd/C », le pourcentage est donné en masse) ou Pd(OH)2/C à 10 ou 20 % (le catalyseur de Pearlman) qui est plus actif dans certains cas. On utilise ce genre de catalyseur à hauteur de 10 % en masse, pour les cas simples.
Dans certains cas, le catalyseur hétérogène est modifié par l'action d'un poison sélectif. Celui-ci transforme l'activité du catalyseur pour le rendre partiellement réactif, afin de n'hydrogéner qu'une ou plusieurs fonctions choisies, sans en affecter d'autres, par exemple l'hydrogénation d'un groupe alcène sans toucher au(x) cycle(s) aromatique(s) aussi présents, ou à l'hydrogénation partielle des alcynes pour former l'alcène correspondant, et pas directement l'alcane (double hydrogénation). C'est par exemple le cas du catalyseur de Lindlar, catalyseur à base de palladium sur du carbonate de calcium, empoisonné par un composé comportant du plomb (acétate de plomb, oxyde de plomb) afin de désactiver une partie des sites du palladium, et de le rendre ainsi moins réactif. Ce catalyseur permet par exemple la conversion du phénylacétylène en styrène[9].
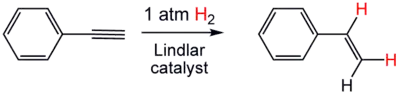 Hydrogénation partielle du phénylacétylène par l'action du catalyseur de Lindlar.
Hydrogénation partielle du phénylacétylène par l'action du catalyseur de Lindlar.
Alors que l'hydrogénation est dans la plupart des cas une addition syn, une hydrogénation asymétrique est également possible par une catalyse hétérogène, sur un métal modifié par un ligand chiral[10].
Mécanisme
Les mécanismes en catalyse hétérogène et dans la plupart des cas en catalyse homogène sont relativement similaires. Le catalyseur (métal) se lie à une molécule de dihydrogène, puis au composé à la liaison multiple, et transfère un à un les atomes d'hydrogène. De par ce mécanisme, les atomes d'hydrogène se retrouvent ajoutés du même côté de la liaison multiple, la réaction est donc une addition syn.
Catalyse hétérogène
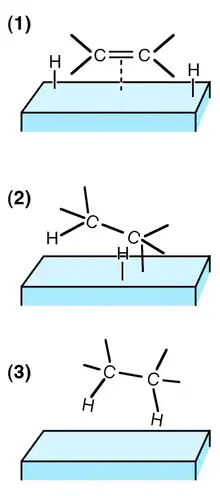
Chaque atome de la molécule de dihydrogène se lie au catalyseur (en général un métal), rendant la liaison entre les deux atomes plus fragile, faisant supporter à chaque atome un déficit en électron, et les rendant ainsi réactif vis-à-vis d'une liaison multiple riche en électrons. Un atome d'hydrogène est additionné à la double liaison (cette étape étant réversible) puis l'autre (cette étape étant alors irréversible dans les conditions de l'hydrogénation).
Catalyse homogène
Dans la plupart des hydrogénations par catalyse homogène[11], le métal se lie au composé et au dihydrogène pour former un intermédiaire qui est un complexe composé-métal-(H)2. On suppose que la séquence générale des réactions est celle décrite ci-dessous, ou du moins en est très proche (on prend ici pour exemple de composé un alcène).
- Liaison entre le catalyseur métallique et le dihydrogène pour former un complexe dihydride (« addition oxydante »), avec au préalable formation d'un complexe dihydrogène :
- LnM + H2 → LnMH2.
- Liaison avec l'alcène :
- LnM(H2) + CH2=CHR → Ln-1MH2(CH2=CHR) + L.
- Transfert de l'un des atomes d'hydrogène du métal au carbone (« insertion migratoire ») :
- Ln-1MH2(CH2=CHR) → Ln-1M(H)(CH2-CH2R).
- Transfert du second atome d'hydrogène du métal au groupe alkyle avec dissociation simultanée de l'alcane (« élimination réductrice ») :
- Ln-1M(H)(CH2-CH2R) → Ln-1M + CH3-CH2R.
Effet de la pression d'hydrogène
En augmentant la pression d'hydrogène, on accroit la solubilisation de ce gaz dans le solvant ce qui, à son tour, augmente le nombre de molécules d'hydrogène dissociées (activées) par le métal. En revanche, si l'on augmente trop la pression d'hydrogène, on peut réduire la solubilité du réactif principal qui peut cristalliser/précipiter sur les particules de catalyseur ; ceci bloque la réaction d'hydrogénation.
Applications
Hydrogénation de liaisons multiples carbone-carbone
Une importante application de l'hydrogénation catalytique en chimie organique est l'hydrogénation de liaisons multiples (double ou triple) carbone-carbone. Cette technique est d'ailleurs utilisée dans l'industrie agroalimentaire, pour saturer les lipides insaturés afin de les rendre solides. On utilise pour cette hydrogénation en général du dihydrogène avec un catalyseur insoluble, comme le palladium, le platine, l'iridium, ou du nickel (nickel de Raney).
L'hydrogénation partielle de la triple liaison des alcynes peut former deux composés, les isomères E ou Z de l'alcène correspondant. Le choix du catalyseur utilisé permet parfois d'obtenir préférentiellement l'un des deux stéréoisomères (exemples : l'utilisation du catalyseur de Lindlar permet de produire préférentiellement l'isomère Z ; le système sodium/ammoniac oriente plutôt vers l'isomère E).
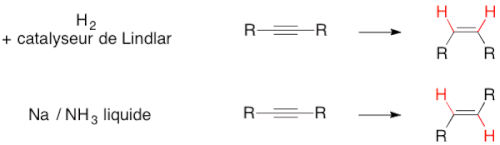
Cependant, dans les conditions de l'hydrogénation, les alcènes sont bien souvent rapidement eux-mêmes hydrogénés en alcane ; en utilisant des catalyseurs spéciaux, moins réactifs, comme le catalyseur de Lindlar, on peut arrêter l'hydrogénation au stade alcène.
L'encombrement stérique joue un rôle dans la facilité de l'hydrogénation catalytiques des alcènes, et permet soit de faire cette réaction à pression et température « normales », soit oblige à des conditions plus drastiques. Ainsi un alcène monosubstitué sera facilement hydrogéné, alors qu'un alcène portant de nombreux substituants sera difficile à hydrogéner.
L'hydrogénation peut aussi être une mesure de stabilité des composés insaturés. Par exemple en observant l'hydrogénation du cyclohexène et celle du cyclohexa-1,3-diène, on peut extrapoler pour déduire la réaction du cyclohaxatriène théorique (avec liaisons doubles localisées) et ainsi comparer sa stabilité avec le benzène, ce qui permet de mesurer l'effet stabilisant produit par la délocalisation des liaisons doubles dans un composé aromatique.
Hydrogénation d'autres liaisons multiples
Il est également possible de réduire par hydrogénation des liaisons multiples carbone-hétéroatome ou hétéroatome-hétéroatome. Il est en particulier possible de réduire les groupes nitro, imine et oxime.
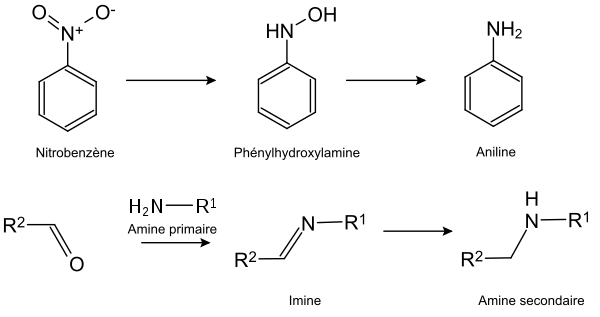
Le procédé Haber est par exemple un procédé à grande échelle effectuant l'hydrogénation du diazote pour produire de l'ammoniac :
![{\displaystyle \overbrace {\ce {N{\equiv }N}} ^{\text{diazote}}\;\;\;+\overbrace {3\,{\color {red}{\ce {H}}}{\ce {_{2}}}} ^{\underset {\text{(200 atm)}}{\text{dihydrogène}}}{\ce {->[{\ce {Fe\ catalyseur}}][350-550\,^{\circ }{\ce {C}}]}}\overbrace {{\ce {2N}}{\color {red}{\ce {H}}}{\ce {_{3}}}} ^{\text{ammoniac}}}](https://img.franco.wiki/i/eb1e9e3f7a1b7d8793929b3e6bca6186a15ee474.svg "Hydrogénation du diazote")
Le dioxygène peut aussi être partiellement hydrogéné en peroxyde d'hydrogène, cependant ce procédé est peu développé.
Déprotection par hydrogénation
En synthèse organique, il est courant de protéger certains groupes fonctionnels, sensibles à des réactions destinées à d'autres parties de la molécule, protection qu'on retire une fois la ou les réactions sensibles effectuées.
En raison des conditions douces dans laquelle elle s'effectue, l'hydrogénation catalytique est un moyen efficace et adapté pour supprimer une protection sur un groupe. Elle est en particulier utilisée pour retirer les groupes protecteurs des groupes benzyle, comme les éthers de benzyle ou les esters et dérivés d'esters de benzyle.
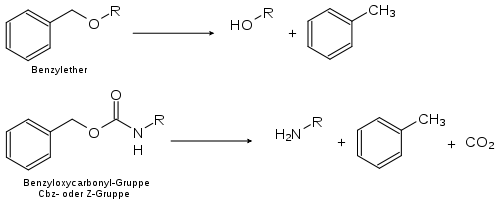
Industrie
L'hydrogénation a de nombreuses applications industrielles. On peut notamment citer le procédé Haber, processus d'hydrogénation du diazote en ammoniac, ou encore le procédé Bergius pour fabriquer de l'essence de synthèse à partir du charbon.
L'hydrogénation est aussi utilisé dans l’industrie pétrochimique, pour convertir les alcènes en alcanes, moins volatils, et les composés aromatiques en alcanes saturés (paraffines) et en cycloalcanes (naphtènes). On peut aussi citer l'hydrocraquage de résidus lourds en gazole.
Le xylitol, un polyol, est produit par hydrogénation du xylose, un aldose.
Industrie agroalimentaire
Elle est entre autres utilisée pour rendre solides des matières grasses originellement liquides (huiles). Certaines margarines sont produites par ce procédé.
Effets sur la santé
L'un des effets secondaires de l'hydrogénation incomplète des matières grasses est l'isomérisation de certains composés insaturés avec en particulier production d'acides gras trans alors que ce sont au contraire les formes cis qui prédominent naturellement. Ceci résulte de la plus faible enthalpie standard de formation des composés trans, dont la formation est ainsi thermodynamiquement favorisée, aboutissant à un ratio d'équilibre cis/trans d'environ 1:2. Cependant, les acides gras trans accroissent les risques de maladies cardio-vasculaires, de sorte que la législation en Europe et aux États-Unis tend à rendre obligatoire l'affichage du contenu en lipides saturés sur les étiquettes des produits vendus dans le commerce et à en restreindre la distribution, notamment au Danemark, en Suisse et à New York.
Références
- (en) K. Amoa, « Catalytic Hydrogenation of Maleic Acid at Moderate Pressures », J. Chem. Educ., vol. 84, no 12, , p. 1948 (ISSN 0021-9584, DOI 10.1021/ed084p1948).
- (en) V. Voorhees et R. Adams, « The use of the oxides of platinum for the catalytic reduction of organic compounds. I », J. Am. Chem. Soc., vol. 44, no 6, , p. 1397–1405 (ISSN 0002-7863, DOI 10.1021/ja01427a021).
- (en) M.-Y. Ngai, J.-R. Kong et al., « Hydrogen-Mediated C−C Bond Formation: A Broad New Concept in Catalytic C−C Coupling », J. Org. Chem., vol. 72, no 4, , p. 1063–1072 (ISSN 0022-3263, DOI 10.1021/jo061895m).
- (en) P. N. Rylander, Ullmann's Encyclopedia of Industrial Chemistry, Weinheim, Germany, John Wiley & Sons, (DOI 10.1002/14356007.a13_487), « Hydrogenation and Dehydrogenation ».
- (en) T. van Es et B. Staskun, « Aldehydes from Aromatic Nitriles: 4-Formylbenzenesulfonamide », Org. Synth., vol. 51, , p. 20 (ISSN 0078-6209, DOI 10.15227/orgsyn.051.0020).
- (en) C. F. H. Allen et J. VanAllan, « m-Toylybenzylamine », Org. Synth., vol. 21, , p. 108 (ISSN 0078-6209, DOI 10.15227/orgsyn.021.0108).
- (en) A. B. Mekler, S. Ramachandran et al., « 2-Methyl-1,3-Cyclohexanedione », Org. Synth., vol. 41, , p. 56 (ISSN 0078-6209, DOI 10.15227/orgsyn.041.0056).
- (en) R. E. Ireland et P. Bey, « 2-Homogeneous Catalytic Hydrogenation: Dihydrocarvone », Org. Synth., vol. 53, , p. 63 (ISSN 0078-6209, DOI 10.15227/orgsyn.053.0063).
- (en) H. Lindlar et R. Dubuis, « Palladium Catalyst for Partial Reduction of Acetylenes », Org. Synth., vol. 46, , p. 89 (ISSN 0078-6209, DOI 10.15227/orgsyn.046.0089).
- (en) T. Mallat, E. Orglmeister et al., « Asymmetric Catalysis at Chiral Metal Surfaces », Chem. Rev., vol. 107, no 11, , p. 4863–4890 (ISSN 0009-2665, DOI 10.1021/cr0683663).
- (en) J. de Vries et J. Cornelis, The Handbook of Homogeneous Hydrogenation, Weinheim, Germany, Wiley-VCH, , 1568 p. (ISBN 978-3-527-31161-3 et 9783527619382, DOI 10.1002/9783527619382).