Addition oxydante
L'addition oxydante et l'élimination réductrice sont deux réactions importantes de la chimie organométallique[1] - [2] - [3] - [4]. L'addition oxydante est un processus qui augmente le degré d'oxydation et le nombre de coordination du centre métallique du complexe qui réagit. Cette réaction est généralement une étape d'un cycle catalytique ; il en est de même de étape inverse : l'élimination réductrice[5]. Il est impossible d'avoir une de ces deux réactions dans un cycle sans que l'autre ne soit présente, car il faut restaurer le degré d'oxydation ainsi que le nombre de coordination du centre métallique.
Rôle dans la chimie des métaux de transitions
Pour les métaux de transitions, les réactions d'oxydation changent le nombre d'électrons de la couche d, généralement par une perte d'électrons : dn devient dn-2. Les additions oxydantes sont favorisées pour des métaux qui satisfont une des deux conditions : (1) être basique et (2) être facilement oxydable. Les métaux avec un degré d'oxydation relativement bas satisfont souvent au moins une de ces deux conditions, mais même certains métaux avec un haut degré d'oxydation peuvent également donner une addition oxydante. Un exemple illustrant cette réaction est l'oxydation de Pt(II) avec le dichlore :
- [PtCl4]2− + Cl2 → [PtCl6]2−
On considère généralement en chimie organométallique que le degré d'oxydation formel du centre métallique et le nombre d'électrons de valence du complexe augmentent tous les deux de deux unités[6]. Cependant, des échanges mono-électroniques sont également possibles et observables dans certaines additions oxydantes. Bien que les réactions d'addition oxydante puissent théoriquement insérer un métal dans une liaison de n'importe quel ligand, dans la pratique, la plupart d'entre elles sont observées avec des ligands de type H–H, H–X, et C–X car ce sont les ligands les plus utilisés pour les applications commerciales de la chimie organométallique.
Une addition oxydante ne peut se faire que si le complexe métallique a une site de coordination vacant. C'est pour cela que les additions oxydantes ne mettent en jeu que des métaux tétra- ou pentacoordonnés.
L'élimination réductrice est la réaction inverse de l'addition oxydante[7]. Elle est favorisée quand la liaison X-Y qui se forme entre les ligands éliminés est forte. Pour que l'on puisse observer une élimination réductrice, les deux groupes (X et Y) doivent être mutuellement adjacents sur la sphère de coordination du métal (on dit parfois que c'est une cis-élimination). Les réactions d'élimination réductrice sont celles qui libèrent les produits dans beaucoup de cycles catalytiques qui forment des liaisons C-H et C-C[5].
Mécanisme de l'addition oxydante
Les réactions d'additions oxydantes sont régies selon différents mécanismes, qui dépendent du centre métallique et des ligands.
Mécanisme concerté
Les additions oxydantes de ligands non polaires tels que le dihydrogène ou les hydrocarbures semblent mettre en jeu un mécanisme concerté. Il se crée une liaison σ à trois centres (le métal, X et Y) vont, de façon simultanée, échanger des électrons π, ce qui dissocie la liaison X-Y, et qui crée les deux nouvelles liaisons X-métal et métal-Y. Ce sont ces nouvelles liaisons σ qui forment le complexe oxydé. On qualifie les nouvelles liaisons de cis[2] bien que l'isomérisation reste possible une fois les liaisons formées. Les autres mécanismes ne conduisent quant à eux généralement pas à un produit cis.
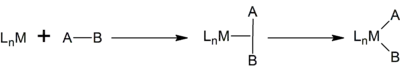
Ce mécanisme est observé lors de l'addition de molécule homonucléaire comme par exemple H2. Beaucoup de liaisons M-(C-H) sont également formées par un mécanisme concerté, selon une interaction agostique[2].
Un exemple assez représentatif est la réaction de l'hydrogène avec le complexe de Vaska, trans-IrCl(CO)[P(C6H5)3]2. Dans cet exemple, l'iridium change son degré d'oxydation initial de +1 à +3. Ce produit est lié à trois anions : un ion chlorure et deux ions hydrures. Comme nous pouvons l'observer en dessous, le complexe initial a 16 électrons et un nombre de coordination de quatre, et le produit a 18 électrons et un nombre de coordination de six.
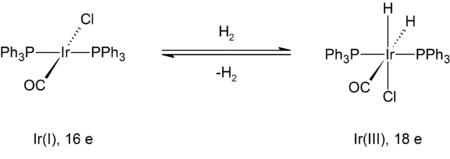
La formation d'un intermédiaire avec le dihydrogène à géométrie moléculaire bipyramidale trigonale est suivie par le clivage de la liaison H-H, causé par un effet de rétrodonation dans l'orbitale σ* de la molécule H-H. Ce système est à l'équilibre chimique. La réaction inverse, faite par une élimination réductrice, donne simultanément une réduction de centre métallique et l'élimination de dihydrogène gazeux[8].
La rétrodonation dans l'orbitale σ* de la liaison H-H est favorisée par un métal riche en électrons[8].
Mécanisme de type SN2
Certaines additions oxydantes ont un procédé analogue à celui d'une substitution nucléophile bimoléculaire en chimie organique. Les attaques nucléophiles du centre métallique sur un atome moins électronégatif du ligand conduit au clivage de la liaison R-X pour former une espèce de la forme [M–R]+. Cette étape est suivie par une coordination rapide de l'anion vers le centre cationique du métal. Un tel mécanisme est observé par exemple lors de la réaction entre un complexe plan carré et l'iodométhane :

Ce mécanisme est souvent observé dans l'addition de ligands polaires et électrophiles, tels que les halogènes et les halogénoalcanes[2].
Mécanisme ionique
Les mécanismes ioniques de l'addition oxydante sont similaires au mécanisme de la SN2, dans la mesure où cela implique l'addition de deux fragments distincts du ligand, en deux étapes successives. La différence entre les deux mécanismes réside dans le fait que le mécanisme ionique implique que le ligand s'est dissocié en deux fragments dans la solution, avant toute interaction avec le centre métallique. L'addition de l'acide chlorhydrique se fait par exemple selon un mécanisme ionique[2].
Mécanisme radicalaire
Les halogénoalcanes et d'autres ligands similaires peuvent s'additionner sur le centre métallique en passant par une forme radicalaire. Certains détails de ce mécanisme restent encore incertains[2], mais il a été accepté que certaines réactions passent par un centre radicalaire. Un exemple a été proposé par Lednor[9].
- Initiation
- [(CH3)2C(CN)N]2 → 2 (CH3)2(CN)C• + N2
- (CH3)2(CN)C• + PhBr → (CH3)2(CN)CBr + Ph•
- Propagation
- Ph• + [Pt(PPh3)2] → [Pt(PPh3)2Ph]•
- [Pt(PPh3)2Ph]• + PhBr → [Pt(PPh3)2PhBr] + Ph•
Applications
Les additions oxydantes et les éliminations réductrices interviennent dans beaucoup de cycles catalytiques. On observe ces réactions dans les réactions avec une catalyse homogène (c'est-à-dire en solution) telles que la réaction de Monsanto, l'hydrogénation des alcènes utilisant le catalyseur de Wilkinson. On suppose également que ces réactions interviennent dans les cycles catalytiques utilisant une catalyse hétérogène tels que l'hydrogénation sur platine. Cependant, dans le cas des catalyses hétérogènes, les métaux son caractérisés par des structures de bandes, le degré d'oxydation n'a donc pas beaucoup de sens.
Les additions oxydantes interviennent aussi dans les additions nucléophiles, ainsi que dans beaucoup de réactions de couplages telles que la réaction de Suzuki, le couplage de Negishi ou le couplage de Sonogashira.
Notes et références
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Oxidative_addition » (voir la liste des auteurs).
- Jay A. Labinger, Tutorial on Oxidative Addition, Organometallics, 2015, volume 34, pp 4784–4795. DOI 10.1021/acs.organomet.5b00565
- (en) Robert Crabtree, The Organometallic Chemistry of the Transition Metals, Wiley-Interscience, , 159–180 p. (ISBN 0-471-66256-9)
- (en) Gary L. Miessler et Donald A. Tarr, Inorganic Chemistry, 3rd
- (en) D. F. Shriver et P. W. Atkins, Inorganic Chemistry
- (en) J. F. Hartwig, Organotransition Metal Chemistry, from Bonding to Catalysis, New York, University Science Books, (ISBN 1-891389-53-X)
- (en) « oxidative addition », IUPAC, Compendium of Chemical Terminology [« Gold Book »], Oxford, Blackwell Scientific Publications, 1997, version corrigée en ligne : (2019-), 2e éd. (ISBN 0-9678550-9-8)
- (en) « reductive elimination », IUPAC, Compendium of Chemical Terminology [« Gold Book »], Oxford, Blackwell Scientific Publications, 1997, version corrigée en ligne : (2019-), 2e éd. (ISBN 0-9678550-9-8)
- (en) Curtis Johnson et Richard Eisenberg, « Stereoselective Oxidative Addition of Hydrogen to Iridium(I) Complexes. Kinetic Control Based on Ligand Electronic Effects », Journal of the American Chemical Society, vol. 107, no 11, , p. 3148–3160 (DOI 10.1021/ja00297a021)
- (en) Thomas L. Hall, Michael F. Lappert et Peter W. Lednor, « Mechanistic studies of some oxidative-addition reactions: free-radical pathways in the Pt0-RX, Pt0-PhBr, and PtII-R′SO2X Reactions (R = alkyl, R′ = aryl, X = halide) and in the related rhodium(I) or iridium(I) Systems », J. Chem. Soc., Dalton Trans., no 8, , p. 1448–1456 (DOI 10.1039/DT9800001448)
Bibliographie
- (en) Valentine P. Ananikov, Djamaladdin G. Musaev et Keiji Morokuma, « Theoretical Insight into the C−C Coupling Reactions of the Vinyl, Phenyl, Ethynyl, and Methyl Complexes of Palladium and Platinum », Organometallics, vol. 24, no 4, , p. 715 (DOI 10.1021/om0490841)
Liens externes
- (en) R. Toreki, « Oxidative Addition », The Organometallic HyperTextBook, Interactive Learning Paradigms Inc.
- (en) R. Toreki, « Reductive Elimination », The Organometallic HyperTextBook, Interactive Learning Paradigms Inc.