Hydrogénation par transfert
L'hydrogénation par transfert est une technique d'hydrogénation dans laquelle la source en hydrogène n'est pas le dihydrogène, mais un autre « donneur en hydrogène ». Ces donneurs sont souvent des solvants comme l'hydrazine, le dihydronaphtalène, le dihydroanthracène, l'isopropanol, l'acide méthanoïque[1] ou le cyclohexadiène. Cette technique est utilisée dans l'industrie et en synthèse organique du fait des inconvénients et des coûts liés à l'utilisation de H2.
En synthèse organique, l'hydrogénation par transfert est utile pour la réduction de composés insaturés polaires comme les cétones, les aldéhydes ou les imines. Une application à grande échelle de l'hydrogénation par transfert est la liquéfaction du charbon en utilisant des « solvants donneurs » tels que la tétraline[2] - [3].
Catalyseurs organométalliques
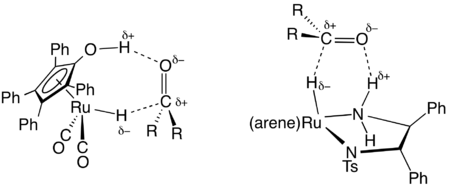
En synthèse organique, une famille utile des catalyseurs transférant de l'hydrogène a été développée à base de complexes de ruthénium et de rhodium, souvent avec des ligands diamine et phosphine[4]. Un important précurseur de catalyseur est dérivé du dimère de dichlorure de (cymène)ruthénium et de diphényléthylènediamine tosylé. Ces catalyseurs sont principalement utilisés pour la réduction de cétones et d'imines en alcools et amines, respectivement. Le donneur d'hydrogène (agent de transfert) est typiquement l'isopropanol qui est converti en acétone après son don d'hydrogène. Les hydrogénations par transfert peuvent être hautement énantiosélectives lorsque le réactif initial est prochiral :
- RR'C=O + Me2CHOH → RR'C*H-OH + Me2C=O
où RR'C*H-OH est un produit chiral. Un catalyseur typique est (cymène)R,R-HNCHPhCHPhNTs, où Ts est le groupe tosyle (SO2C6H4Me) et R,R est la configuration absolue des deux carbones chiraux. Les travaux de Ryōji Noyori sur ce type de réaction lui ont valu le prix Nobel de chimie en 2001[5].
Une autre famille d'agent transférateur d'hydrogène sont les alcoolates d'aluminium, tels que l'isopropanolate d'aluminium dans la réduction de Meerwein-Ponndorf-Verley ; cependant leur activité est relativement faible en comparaison avec les composés à base de métaux de transition.
Synthèses sans métal
Avant l'avènement de l'hydrogénation catalytique, de nombreuses méthodes furent développées pour hydrogéner les composés insaturés. La plupart de ces méthodes n'ont aujourd'hui d'intérêt qu'historique ou pédagogique. Un des principaux agents de transfert de ces réactions est le diimide, qui est oxydé pour donner le diazote (N2), composé particulièrement stable.
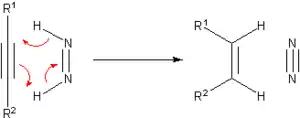
Le diimide est lui-même produit à partir de l'hydrazine. On peut citer parmi le hydrocarbures pouvant servir de source en hydrogène le cyclohexène et le cyclohexadiène. Dans ce cas, en plus du composé hydrogéné, il se forme un équivalent de benzène, la principale force favorisant cette réaction étant la baisse d'énergie/augmentation de la stabilisation du fait de la production d'un composé aromatique. Du palladium peut être utilisé pour catalyser cette réaction, qui peut alors se produire à 100 °C.
Des hydrogénations par transfert plus particulières ont également été rapportées. Parmi celles-ci, l'hydrogénation intramoléculaire ci-dessous :
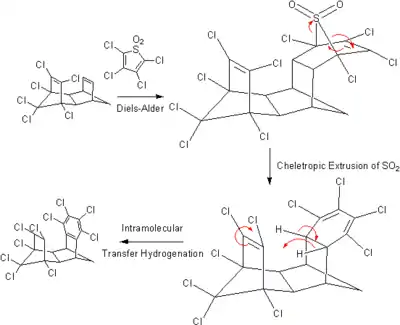
Dans cette réaction, le 1,8,9,10,11,11-hexachlorotétracyclo[6.2.1.13,6.02,7]dodéca-4,9-diène (à gauche), qui joue ici le rôle de diénophile, va d'abord réagir avec le 1,1-dioxyde de 2,3,4,5-tétraclorothiophène (diène) par réaction de Diels-Alder. L'adduit va ensuite procéder à une éjection chélétrope de dioxyde de soufre, formant un composé possédant à présent à son extrémité droite un groupe tétrachlorocyclohexadiène, dont la liaison saturée fait face à la seconde liaison double du composé initial. On observe alors un transfert des deux atomes d'hydrogène de l'une à l'autre, la réaction formant un groupe tétrachlorobenzène bien plus favorable énergétiquement.
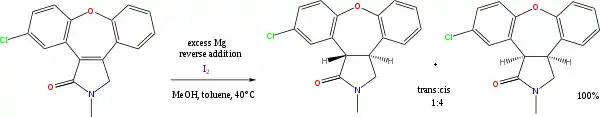
De nombreuses réactions existent avec un alcool ou une amine dans le rôle de donneur de proton, et un métal alcalin dans celui de donneur d'électron. Une de ces réactions qui continue aujourd'hui de présenter un intérêt est la réduction de Birch des arènes en présence de sodium métallique. Une autre, moins importante, est la réduction de Bouveault et Blanc des esters. Enfin, la combinaison magnésium/méthanol est aussi utilisée pour la réduction d'alcènes, par exemple dans la synthèse de l'asénapine[6] :
Hydrogénation par transfert organocatalytique
L'hydrogénation par transfert organocatalytique a été décrite par le groupe de List en 2004, dans un système utilisant un ester de Hantzsch comme donneur d'hydrure et une amine comme catalyseur[7] :
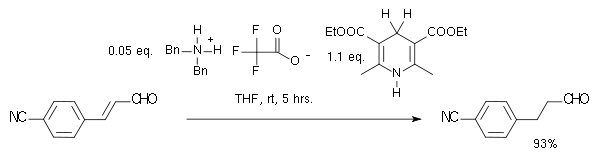
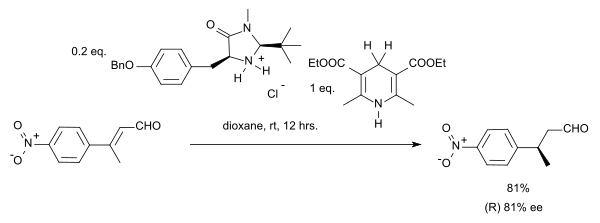
Dans cette réaction, le substrat est un composé carbonylé α,β-insaturée. Le donneur de proton est oxydé en une forme de type pyridine ressemblant à la coenzyme NADH. Dans le cycle catalytique de cette réaction, l'amine et l'aldéhyde forment d'abord un ion iminium, puis le transfert de proton est suivi par une hydrolyse de l'iminium régénérant le catalyseur. En utilisant un organocatalyseur de MacMillan (imidazolidinone chirale), une énantiosélectivité de 81% ee a été obtenue :
Le groupe de MacMillan a publié indépendamment une synthèse asymétrique très similaire en 2005[8] :
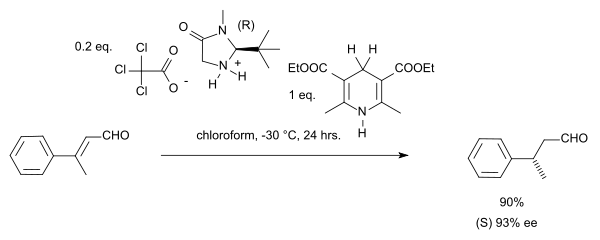
Cette réaction présente un intéressant cas de stéréoconvergence, les isomères E ou Z produisant finalement chacun l'énantiomère S.
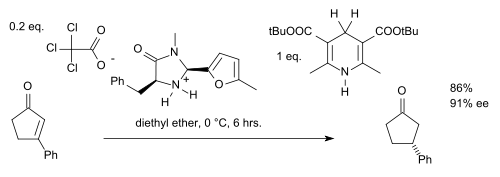
Pour étendre ce type de réaction aux cétones, ou plutôt aux énones, il faut ajuster précisément le catalyseur (ajouter un groupe benzyle et remplacer le groupe tert-butyle par un furane) ainsi que l'ester de Hantzsch (rajouter de plus de volumineux groupes tert-butyle)[9] :
Avec des organocatalyseurs totalement différents, il est possible d'hydrogéner des imines. Un exemple de ce type de réaction est l'hydrogénation d'une quinoléine, catalysée par un acide phosphorique dérivé du BINOL, pour former une tétradéshydroquinoléine chirale. Cette synthèse est une réaction en cascade, l'enchaînement d'une addition 1,4, d'une isomérisation et d'une addition 1,2[10] :
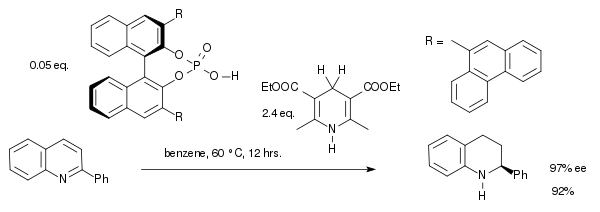
La première étape de la réaction est la protonation de l'atome d'azote de la quinoléine par l'acide phosphorique, formant un intermédiaire iminium chiral. Il a été noté qu'avec des catalyseurs métalliques plus traditionnels, l'hydrogénation de composés aromatiques ou hétéroaromatiques a tendance à échouer.
Notes et références
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Transfer hydrogenation » (voir la liste des auteurs).
- (en) T. van Es et B. Staskun, « Aldehydes from Aromatic Nitriles: 4-Formylbenzenesulfonamide », Org. Synth., vol. 51, , p. 20 (ISSN 0078-6209, DOI 10.15227/orgsyn.051.0020).
- (en) Speight, J. G., The Chemistry and Technology of Coal, New York, Marcel Dekker, (ISBN 0-8247-1915-8), p. 226
- Kilian Muñiz, « Bifunctional Metal-Ligand Catalysis: Hydrogenations and New Reactions within the Metal-(Di)amine Scaffold13 », Angewandte Chemie International Edition, vol. 44, no 41, , p. 6622–6627 (PMID 16187395, DOI 10.1002/anie.200501787)
- (en) T. Ikariya, K. Murata, R. Noyori, « Bifunctional Transition Metal-Based Molecular Catalysts for Asymmetric Syntheses », Org. Biomol. Chem., vol. 4, , p. 393-406
- Shimizu, H., Nagasaki, I., Matsumura, K., Sayo, N., and Saito, T, « Developments in Asymmetric Hydrogenation from an Industrial Perspective », Acc. Chem. Res., vol. 40, , p. 1385-1393 (DOI 10.1021/ar700101x)
- M. V. D. Linden, T. Roeters, R. Harting, E. Stokkingreef, A. S. Gelpke et G. Kemperman, « Debottlenecking the Synthesis Route of Asenapine », Organic Process Research & Development, vol. 12, no 2, , p. 196–201 (DOI 10.1021/op700240c)
- Yang, M. Hechavarria Fonseca et B. List, « A metal-free transfer hydrogenation: organocatalytic conjugate reduction of alpha,beta-unsaturated aldehydes », Angewandte Chemie (International ed. in English), vol. 43, no 48, , p. 6660–6662 (PMID 15540245, DOI 10.1002/anie.200461816)
- Ouellet, J. Tuttle et D. MacMillan, « Enantioselective organocatalytic hydride reduction », Journal of the American Chemical Society, vol. 127, no 1, , p. 32–33 (PMID 15631434, DOI 10.1021/ja043834g)
- Tuttle, S. Ouellet et D. MacMillan, « Organocatalytic transfer hydrogenation of cyclic enones », Journal of the American Chemical Society, vol. 128, no 39, , p. 12662–12663 (PMID 17002356, DOI 10.1021/ja0653066)
- Rueping, A. Antonchick et T. Theissmann, « A highly enantioselective Brønsted acid catalyzed cascade reaction: organocatalytic transfer hydrogenation of quinolines and their application in the synthesis of alkaloids », Angewandte Chemie (International ed. in English), vol. 45, no 22, , p. 3683–3686 (PMID 16639754, DOI 10.1002/anie.200600191)