Mitochondrie
Une mitochondrie est un organite, possédant toutes les caractéristiques d'un organisme procaryote, entourée d'une double membrane composée chacune d'une double couche phospholipidique, et retrouvée chez la plupart des cellules eucaryotes[1] (absente dans les érythrocytes matures et chez certains parasites). Leur diamètre varie généralement entre 0,75 et 3 µm tandis que leur forme générale et leur structure sont extrêmement variables[2]. On en retrouve jusqu'à 2 000 par cellule, et elles sont localisées préférentiellement au niveau des zones cellulaires consommatrices d'adénosine triphosphate (ATP). Elles permettent la production d'ATP, de divers cofacteurs métaboliques (NADH, FADH2) et sont impliquées dans différents processus tels que la communication, la différenciation, l'apoptose et la régulation du cycle cellulaire[3]. Les mitochondries sont aussi liées à certaines maladies humaines telles que des retards mentaux[4], des problèmes cardiaques[5] et jouent un rôle important dans le processus de vieillissement.
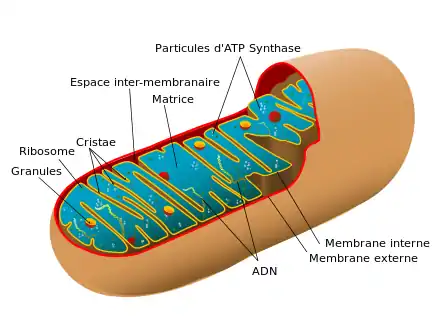
Les mitochondries sont invisibles en microscopie optique lorsqu'elles ne sont pas teintées par des colorants biologiques (rhodamine 123 et vert Janus B). Leur étude détaillée fait appel au microscope électronique qui possède une bien meilleure résolution.
La théorie endosymbiotique explique[6] la présence de mitochondries dans les cellules eucaryotes par l'incorporation ou endocytose d'une α-protéobactérie dans une cellule hôte, il y a plusieurs milliards d'années. L'ADN des mitochondries est ainsi différent de celui du noyau, et transmis généralement par la mère.
Étymologie
Le terme mitochondrie provient du grec ancien μίτος (mitos) qui veut dire « fil » et χόνδρος (chondros) qui signifie « granule ».
Fonction
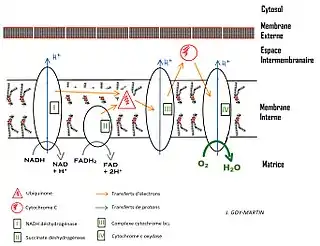
Les mitochondries sont souvent décrites comme les « centrales énergétiques » des cellules, dans la mesure où elles contribuent à l'essentiel de la production d'ATP cellulaire à travers la β-oxydation, le cycle de Krebs et la chaîne respiratoire dans le cadre de la phosphorylation oxydative, l'ATP étant la molécule énergétique ubiquitaire utilisée dans un très grand nombre de réactions chimiques du métabolisme, et notamment de l'anabolisme (biosynthèses). Outre leur rôle dans le métabolisme énergétique cellulaire, les mitochondries interviennent également dans la signalisation, la différenciation et la mort des cellules, ainsi que dans le contrôle du cycle cellulaire et de la croissance de la cellule[7]. Ces processus influencent en retour la biogenèse des mitochondries[8] - [9]. Elles ont par ailleurs été associées à plusieurs maladies humaines, comme des maladies mitochondriales[10] et diverses cardiopathies[5] - [11].
Plusieurs propriétés des mitochondries en font des organites particuliers. Leur nombre par cellule varie considérablement par espèce, par tissu et par type cellulaire (l'ensemble des mitochondries est appelé le chondriome). Ainsi, les globules rouges du sang (hématies) sont totalement dépourvus de mitochondries. Les plaquettes en contiennent très peu, tandis que les cellules du foie et les cellules musculaires peuvent en contenir plus de 2 000. Cet organite est composé de plusieurs compartiments spécialisés dans plusieurs fonctions physiologiques : la membrane mitochondriale externe, l'espace intermembranaire mitochondrial, la membrane mitochondriale interne, et la matrice mitochondriale. Les protéines mitochondriales dépendent des espèces et des tissus considérés. Chez l'homme, les mitochondries cardiaques contiennent au moins 615 types de protéines différents[12], tandis qu'on en a identifié 940 chez le rat[13] ; le protéome mitochondrial est régulé de façon vraisemblablement dynamique[14].
Enfin, les mitochondries possèdent leur propre génome, dit génome mitochondrial, dont l'ADN présente de nombreuses analogies avec le génome des bactéries[15].


Structure

1 : membrane interne,
2 : membrane externe,
3 : espace intermembranaire,
4 : matrice.
On rencontre environ 300 à 2 000 mitochondries par cellule[16]. Les mitochondries y ont un diamètre de 0,75 à 3 μm et une longueur pouvant atteindre 10 µm. Elles se composent de deux membranes, une membrane mitochondriale externe et une membrane mitochondriale interne, qui délimitent trois milieux : le milieu extra-mitochondrial (cytoplasme de la cellule), l'espace intermembranaire mitochondrial, et la matrice mitochondriale.
Membrane externe
La membrane mitochondriale externe contient l'ensemble de l'organite et a une épaisseur d'environ 6 à 7,5 nm. Son rapport massique protéines/phospholipides est semblable à celui des membranes plasmiques des cellules d'eucaryotes, généralement voisin de 1:1.
Elle contient un grand nombre de protéines membranaires intégrales appelées porines et formant des canaux aqueux permettant aux molécules hydrophiles de moins de 5 kDa de diffuser librement à travers la bicouche lipidique[17] : anions, cations, acides gras, nucléotides. La membrane est cependant imperméable aux ions H⁺.
Des protéines, plus massives, peuvent pénétrer dans la mitochondrie lorsqu'une séquence signal est attachée à leur extrémité N-terminale, permettant à ces protéines de se lier à une translocase de la membrane externe, laquelle assure leur transport actif à travers cette membrane[18].
La membrane externe contient également des enzymes impliquées dans des activités aussi diverses que la biosynthèse des acides gras (entrant notamment dans la constitution de la plupart des lipides), l'oxydation de l'adrénaline (une hormone et un neurotransmetteur) et la dégradation du tryptophane (un acide aminé protéinogène). Il s'agit notamment de la monoamine oxydase, de la NADH-cytochrome c réductase insensible à la roténone, de la kynurénine 7,8-hydroxylase et de l'acyl-CoA synthétase.
La rupture de la membrane externe permet aux protéines de l'espace intermembranaire mitochondrial de se répandre dans le cytosol, conduisant à la mort cellulaire[19]. Il s'agit notamment du cytochrome C.
La membrane mitochondriale externe peut s'associer à la membrane du réticulum endoplasmique en une structure désignée par l'abréviation MAM (mitochondria-associated ER-membrane). Cette structure joue un rôle important dans certaines voies de signalisation cellulaire du calcium et intervient dans le transfert de lipides entre le réticulum endoplasmique et les mitochondries[20].
Espace intermembranaire
L'espace intermembranaire mitochondrial, parfois appelé espace périmitochondrial, est délimité par les membranes mitochondriales externes et internes. Dans la mesure où la membrane externe est perméable aux petites molécules, la concentration d'espèces chimiques telles que les oses et certains ions est essentiellement la même dans l'espace intermembranaire que dans le cytosol.
Du fait de l'imperméabilité de la membrane externe aux ions H⁺, l'espace inter-membranaire est saturé de protons provenant des processus métaboliques ayant lieu dans la matrice. Des procaspases et des cytochromes c[19], impliqués dans l'apoptose, sont présents en quantité notable dans l'espace inter-membranaire.
Les protéines qui portent une séquence spécifique de signalisation peuvent être transportées à travers la membrane externe, de sorte que la composition en protéines diffère dans l'espace intermembranaire par rapport à celle du cytosol.
Morphologie et composition
La membrane mitochondriale interne est structurée en crêtes caractéristiques de la mitochondrie, invaginations lamellaires et tubulaires dirigées vers l'intérieur de la matrice et observables en microscopie électronique ou en microscopie par hybridation in situ en fluorescence[21].
Elles abritent les enzymes de la chaîne respiratoire, l'ATP synthase, des perméases, et les chaînes du transport des électrons[22].
La morphologie des crêtes est sensiblement dépendante de la présence d'ATP synthase, laquelle assure l'approvisionnement en ATP de la cellule ; le nombre et la forme des crêtes varie selon l'activité de la mitochondrie (besoins énergétiques importants, oxydation des acides gras)[23] - [24].
Elle est constituée de 3/4 de protéines et 1/4 de lipides. Sa surface est jusqu'à 3 fois plus grande que celle de la membrane externe, du fait des crêtes. La membrane interne contient plus de 151 polypeptides différents, elle héberge environ 1⁄8 de toutes les protéines de la mitochondrie. De ce fait, la concentration en lipides est moindre que celle de la bicouche externe et sa perméabilité est moindre[25] - [26].
La membrane interne possède notamment un phospholipide double, la cardiolipine[27], substituée par quatre acides gras. La cardiolipine est généralement caractéristique des membranes mitochondriales et des membranes plasmiques bactériennes. Dans le corps humain, elle est présente majoritairement dans les régions à haute activité métabolique ou à haute activité énergétique, comme les cardiomyocytes contractiles, dans le myocarde[28] - [21].
D'une bicouche de composition différente, majoritairement protéique, les molécules, les ions et les complexes protéiques passent majoritairement au travers de transporteurs membranaires. Ainsi, des protéines sont transportées par les complexes translocase de la membrane interne (en) (TIM) ou par la protéine Oxa1[18] - [29].
Fonction et métabolisme
Au contraire de la membrane externe, elle ne contient pas de porines, mais des perméases, assurant le co-transport des ions H+ et des molécules.
Ainsi, des protéines sont transportées par les complexes translocase de la membrane interne (en) (TIM) ou par la protéine Oxa1[18]. Ainsi, le complexe TIM 23 permet l'entrée de protéines situées dans l'espace intra-membranaire dans la membrane interne et dans la matrice mitochondriale. Le complexe TIM 22 permet l'insertion des protéines dans la membrane interne et notamment des protéines à plusieurs domaines transmembranaires. Les protéines Oxa permettent la sortie de la matrice pour certaines protéines d'origine mitochondriale.
Les protéines de la membrane mitochondriale interne assurent de nombreuses fonctions physiologiques :
- L'antiport ANT (Adenine Nucleotide Translocase), il fait passer l'ATP de la matrice vers l'espace inter-membranaire et l'ADP de l'espace inter-membranaire vers la matrice ;
- Le transporteur pyruvate-protons. Ces deux éléments diffusent dans le même sens, il s'agit donc d'un symport ;
- Le symport phosphate-protons ;
- Le symport acide gras-protons ;
- La Fo-F1 ATPase faisant passer les protons de l'espace inter-membranaire à la matrice. Ce passage permet la production d'ATP ;
- Le transporteur UCP-1 venant s'ajouter en complément de la Fo-F1 dans les cellules de la graisse brune. UCP-1 amène les protons de l'espace inter-membranaire à la matrice tout comme la Fo-F1. Cependant le passage de protons produira de la chaleur au lieu d'ATP. En effet, l'adipocyte brun produit de la chaleur, il est surtout présent chez les animaux qui hibernent. Le nouveau-né humain en possède également, il perdra ces cellules avec la croissance. Finalement, à l’age adulte, il ne restera que quelques adipocytes bruns au niveau cervical ;
- réactions d'oxydoréduction de la phosphorylation oxydative ;
- production d'ATP à partir d'ADP par l'ATP synthase ;
- transporteurs membranaires assurant et régulant la circulation des métabolites de et vers la matrice mitochondriale ;
- système d'importation des protéines ;
- dynamique mitochondriale : fusion (en) et fission des mitochondries (en), l'équilibre entre ces deux processus étant probablement un déterminant majeur du nombre de mitochondries, de leur longueur et de l'importance de leurs interconnexions[30].
Matrice
La matrice mitochondriale est l'espace inclus dans la membrane mitochondriale interne. Elle renferme environ les deux tiers du total des protéines de la mitochondrie. Elle joue un rôle déterminant dans la production d'ATP avec l'aide de l'ATP synthase incluse dans la membrane interne. Elle contient un mélange très concentré de centaines d'enzymes différentes (principalement impliquées dans la dégradation des acides gras et du pyruvate), de ribosomes spécifiques aux mitochondries, d'ARN de transfert et plusieurs copies de l'ADN du génome mitochondrial.
Les mitochondries possèdent leur propre génome ainsi que l'équipement enzymatique nécessaire pour réaliser leur propre biosynthèse des protéines. La séquence du génome mitochondrial humain se compose de 16 569 paires de bases encodant 37 gènes[31] : 22 ARN de transfert, 2 ARN ribosomique et 13 polypeptides. Les 13 peptides mitochondriaux humains sont intégrées à la membrane mitochondriale interne avec des protéines encodées par des gènes situés dans le noyau de la cellule.
Origine
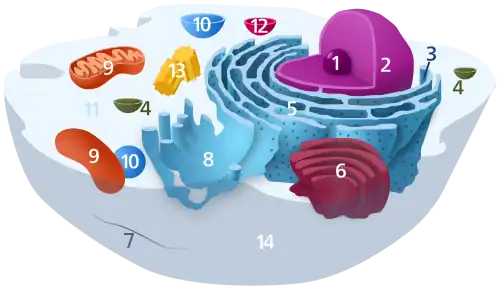
1. Nucléole ;
2. Noyau ;
3. Ribosomes ;
4. Vésicule ;
5. Réticulum endoplasmique rugueux (ou granuleux) (REG) ;
6. Appareil de Golgi ;
7. Cytosquelette ;
8. Réticulum endoplasmique lisse ;
9. Mitochondries ;
10. Vacuole ;
11. Cytosol ;
12. Lysosome ;
13. Centrosome (constitué de deux centrioles) ;
14. Membrane plasmique.
Une étude suggère qu'une symbiose entre Asgardarchaeota, hétérotrophes et rejetant de l'hydrogène ainsi que d'autres composés réduits, et α-protéobactéries spécialisées dans le métabolisme de l'hydrogène aurait précédée l'endosymbiose[33].
Génome mitochondrial
Selon la théorie endosymbiotique, les mitochondries possèderaient une origine monophylétique unique. Une cellule de procaryote primitive aurait intégré un endosymbiote il y a environ 1,5 à 2 milliards d’années, lorsque l’atmosphère primitive s’est enrichie en oxygène[34] - [35]. Les études phylogénétiques indiquent que cet endosymbiote est apparenté aux alphaprotéobactéries, le plus proche parent de la mitochondrie connu actuellement étant Rickettsia prowazekii, un parasite intracellulaire obligatoire[34] (c'est-à-dire une bactérie ne pouvant survivre, se développer et se reproduire qu'à l'intérieur des cellules de son hôte, en utilisant les ressources de ces dernières). Au cours de l’évolution, la majorité des gènes de l’endosymbiote originel auraient été perdus ou bien transférés vers le noyau de la cellule d'eucaryote hôte[35] - [36]. En effet, les nombreux pseudogènes mitochondriaux présents dans le génome attestent d’un processus de transfert tout au long de l’évolution[37] - [38].
Le matériel génétique (ADN mitochondrial) de la mitochondrie (qui est la seule partie des cellules animales à posséder son propre ADN, en plus du noyau) est souvent utilisé dans les recherches phylogénétiques. Le génome mitochondrial (ADNmt) humain est circulaire, ne possède pas d'introns, et est composé de 16 569 paires de bases (génome de petite taille), dont 13 cistrons codant des ARN messagers, 22 gènes codant des ARN de transfert, et 2 gènes codant des ARN ribosomiques.
Le génome mitochondrial peut être très différent d'une espèce à l'autre, il est extrêmement dynamique, et est souvent hétéroplasmique, c'est-à-dire que différentes formes coexistent au sein de la même cellule. Il peut être trouvé sous forme circulaire ou linéaire, double ou simple brin. Ce génome présente 5 à 10 copies dans la mitochondrie. Ces différentes formes sont, entre autres, les produits de la réplication du génome mitochondrial par un mécanisme de cercle roulant, mais aussi d'un mécanisme de réplication recombinaison-dépendant, similaire à la réplication du phage T4. Les génomes mitochondriaux sont habituellement représentés sous forme circulaire, le « cercle maître » qui correspond à la molécule décrivant le mieux le génome.
Les ribosomes mitochondriaux ou mitoribosomes sont différents des ribosomes de la cellule : ils sont plus petits (70S au lieu de 80S).
Le code génétique employé pour la synthèse des protéines peut être différent de celui utilisé dans les synthèses cytosoliques. Chez les vértébrés 4 codons sur 64 ont une signification différente, dont le codon UGA qui est transcrit dans le cytosol en codon stop mais dans la matrice UGA est transcrit en tryptophane (Trp/W), AGG et AGA codent un codon STOP au lieu d'une arginine (Arg/R) et AUA code la méthionine (Met/M) au lieu de l'isoleucine (Ile/I). L'ADN mitochondrial peut aussi se répliquer.
Chez les animaux, lors de la reproduction sexuée, les mitochondries du spermatozoïde pourraient passer dans l'ovocyte, mais le nombre de mitochondries ainsi transférées reste très faible en comparaison de celles déjà présentes dans l'ovocyte. Les mitochondries du spermatozoïde restent localisées sur le flagelle qui sera détruit par autophagie lorsque le spermatozoïde sera dans l'ovocyte. Autrement dit, la quasi-totalité des mitochondries de la cellule-œuf provient du gamète femelle. L'étude de l'ADN mitochondrial humain permet donc de retracer les relations généalogiques entre les individus seulement selon la voie maternelle. Certaines études ont ainsi pu décrire un génome mitochondrial ancestral duquel descendraient tous les génomes mitochondriaux de l'humanité. L'individu femelle supposé qui portait ce génome a été dénommé Ève mitochondriale. Ce terme biblique reste toutefois trompeur, il est en effet très peu probable que l'humanité ait un unique ancêtre féminin et de récentes études, prouvant le transfert de mitochondries provenant des spermatozoïdes lors de la fécondation, remettent en cause cette théorie[39] - [40].
Le code génétique de la mitochondrie est différent de celui du noyau. En effet, le codon AUA code une isoleucine dans le noyau et une méthionine dans la mitochondrie. Le codon UGA est un codon stop (qui arrête la traduction) mais code du tryptophane dans la mitochondrie.
Chez les plantes vertes, l'ADN mitochondrial est bien plus grand et de taille très variable, il code une soixantaine de protéines connues, même si, chez les plantes comme chez les animaux, la vaste majorité des protéines mitochondriales est codée dans le génome nucléaire. Le génome mitochondrial ainsi que le génome chloroplastique contiennent des introns de type II (group-II introns). Les introns de type-II possèdent une origine évolutive commune[41] avec le splicéosome. Ces introns de type-II possèdent une séquence qui a dégénéré au cours de l’évolution et beaucoup ont perdu la faculté de s'auto-épisser de manière indépendante. Ils ont besoin de facteurs codés dans le noyau pour être épissés et parfois également de facteurs codés à l’intérieur de ces organites (appelés les maturases).
Protéome mitochondrial
Le protéome mitochondrial est l'ensemble des protéines présentes dans les mitochondries d'une cellule eucaryote à un moment donné. Le protéome est un ensemble dynamique défini dans le temps (moment considéré : stade de développement, matin ou soir) et dans l'espace (échantillon considéré : cellule, tissu, organisme). Pour décrire l'ensemble des protéines pouvant être présentes dans une mitochondrie à un moment quelconque de la vie de l'organisme, on utilisera le terme de protéome total.
Le protéome mitochondrial est composé de protéines produites dans les mitochondries et codées dans le génome mitochondrial, et de protéines produites dans le cytoplasme et codées dans le génome nucléaire. La plupart des complexes enzymatiques (exemple : ATP-synthase) sont formés par la juxtaposition de polypeptides synthétisés dans la mitochondrie et dans le cytosol (le fluide interne de la cellule).
Bien que les mitochondries soient les descendantes de bactéries, les protéines de leur protéome ne sont pas toutes d'origine bactérienne. Ainsi, chez la levure 50 à 60 % des protéines mitochondriales ont des homologues chez les procaryotes alors que 40 à 50 % n’en ont pas[35].
Il est intéressant de noter que c'est grâce à des associations des protéines de kinésine et de dynéine à des microtubules que la mitochondrie est capable de mouvement.
Protéines mitochondriales codées par le génome mitochondrial
Suivant les organismes 1 à 10 % des protéines mitochondriales sont directement synthétisées dans la matrice par les mitoribosomes, à partir de l'ADN mitochondrial.
Protéines mitochondriales codées par le génome nucléaire
Les protéines mitochondriales possédant un homologue procaryote résultent probablement du transfert des gènes de l’endosymbiote vers le nucléaire tandis que les protéines non homologues à des protéines procaryotes résultent d’un phénomène « d’enrichissement » du protéome mitochondrial par de nouvelles protéines et donc de nouvelles fonctions[34].
Les protéines mitochondriales codées par le génome nucléaire (ou protéines mitochondriales nucléaires) sont importées à l'intérieur de la matrice mitochondriale par différents mécanismes possibles :
- des complexes d'importation (3 sur la membrane interne, 2 sur la membrane externe) ; TOM (Transporter Outer Membrane) est le complexe d'importation situé sur la membrane externe et TIM (Transporter Inner Membrane) est le complexe d'importation situé sur la membrane interne ;
- un peptide signal (environ 15 à 30 acides aminés) de la protéine qui permet sa reconnaissance et son importation dans la mitochondrie[42] - [43] ; il existe des signal-peptidase qui clivent certains peptides signaux, particulièrement ceux situés sur le côté N-terminal ;
- grâce à un apport énergétique.
La différence de potentiel de part et d'autre de la membrane peut provoquer le passage des protéines dans la matrice.
Chez l'homme
La taille du protéome mitochondrial humain est estimée à plus d’un millier de protéines, dont environ 1 % codées par le génome mitochondrial (13 protéines)[44], dont actuellement la moitié est identifiée[45] - [46]. Seules 13 protéines sont codées par l’ADN mitochondrial, vestige du génome de l’endosymbionte. Toutes les autres protéines sont codées par le génome nucléaire.
Fonctionnement
Elle est considérée comme la « centrale énergétique » de la cellule, car c'est là que se déroulent les dernières étapes du cycle respiratoire qui convertit l'énergie des molécules organiques issues de la digestion (glucose) en énergie directement utilisable par la cellule (l'ATP). En cas d'absence d'oxygène la cellule utilise la fermentation dans le cytoplasme pour produire l'énergie nécessaire à son fonctionnement, mais c'est un système bien moins efficace, qui dégrade le substrat de façon incomplète. L'augmentation de la concentration en ions H+ dans les cellules musculaires est une des raisons de la fatigue après une activité intense. En effet, ces ions H+ changent le pH intracellulaire et modifient de fait les conditions de fonctionnement enzymatiques de la cellule qui ne peut plus travailler correctement.
C'est dans la mitochondrie que se déroulent les deux dernières phases de la respiration cellulaire : le cycle de Krebs (dans la matrice) et la chaîne de transport d'électrons (au niveau de la membrane interne). En effet, la production d'ATP comporte 3 principales étapes :
- La glycolyse est la première étape. Elle se déroule dans le cytoplasme cellulaire.
- La deuxième étape est la production d'Acétyl-CoA dans la mitochondrie.
- La troisième et dernière étape est la phosphorylation oxydative.
Au cours de ces 3 étapes, via le cycle de Krebs (donc en condition d'aérobiose), la mitochondrie permet, à partir d'une molécule de glucose, la production théorique de 36 ou 38 molécules d'ATP (cela dépend de la navette utilisée pour transporter le NAD de la glycolyse) — en pratique, le rendement est un peu moins élevé, voisin d'une trentaine de molécules d'ATP par molécule de glucose oxydée, certaines études donnant la valeur de 29,85 ATP/glucose[47].
L'Acétyl-CoA peut aussi être obtenues par transformation de l'acétoacétyl-CoA issu de la transformation de corps cétoniques produits par le foie à partir d'acides gras (jeûne, régime cétonique). Dans le cas du cerveau, cette filière d'apport énergétique présente l'avantage de passer la barrière hémato-encéphalique sans adjuvant (insuline ou protéines spécifiques) pouvant la modifier à terme et pourrait éviter des mécanismes d'inflammations consécutifs à la mauvaise qualité de l'alimentation [48]. De plus, l'apport en énergie est plus rapide et plus efficace (meilleur disponibilité, pas de glycolyse)[49].
Les mitochondries participent à l'apoptose (mort cellulaire) avec le cytochrome c. De plus, elles ont aussi une fonction de concentration et de stockage des ions calcium, sodium et potassium où ils sont stockés sous forme de granules opaques. On trouve également de l'or, du fer et de l'osmium.
Température
Les résultats d'une étude de chercheurs de l'INSERM publiée en 2018[50] - [51] suggèrent que les mitochondries peuvent approcher une température de 50 °C[52], soit au moins 10° de plus que la température de l'organisme, ce qui pourrait ouvrir des pistes prometteuses pour lutter contre certaines maladies[53]. Cependant, les hétérogénéités de température à l'intérieur de la cellule sont habituellement considérées comme impossibles, de précédentes études ayant suggéré un écart de température entre la cellule et les mitochondries d’à peine un millième de degré[54]. Une lecture critique de l'étude de l'INSERM suggère que l'existence d'une différence de température de 10 °C à travers quelques microns entrainerait une consommation d'énergie déraisonnable pour la cellule[55]. Enfin, ces résultats controversés pourraient être dus à une mauvaise interprétation de la sonde moléculaire fluorescente dont le degré de fluorescence dépend de la température mais aussi d'un mouvement[56].
Poisons mitochondriaux
| Cibles des poisons | Poisons |
|---|---|
| Complexe I | roténone, barbituriques, dérivés mercuriels |
| Complexe II | malonate (acide malonique), SDHI (pesticide) |
| Complexe III | antimycine, strobilurine (pesticide) |
| Complexe IV | monoxyde d'azote, cyanure, monoxyde de carbone |
| Complexe V (F0/F1ATPase) | oligomycine, aurovertine |
| Échangeur ATP/ADP | atractyloside, acide bongkrékique (en) |
| Perméabilité de la membrane interne | dinitrophénol, valinomycine |
Dans l'agriculture, la mitochondrie est la cible préférée des pesticides, la roténone tout d'abord, utilisée puis interdite car elle est reliée à la maladie de Parkinson. Actuellement ce sont les SDHI, des inhibiteurs de la succinate déshydrogénase qui sont très utilisés dans le but d'éliminer les moisissures. Certains poisons ont pour rôle non pas d'empêcher les différents complexes de fonctionner, c'est-à-dire que les transferts d'électron de la chaîne respiratoire sont effectués mais ces protéines, les découplants ou UCP vont court-circuiter le complexe V (ATP synthase) en créant un canal à travers la membrane interne. Ce pore permet aux protons de passer de l'espace intermembranaire vers la matrice dans le sens de leur gradient, ce qui se traduit par un dégagement de chaleur mais aucune production d'ATP. On peut citer ici l'exemple du dinitrophénol.
Maladies mitochondriales
Les mitochondries, dès le stade fœtal ont rôle essentiel (notamment pour le métabolisme intermédiaire, le développement neurologique périnatal, l'immunité, la bioénergétique, le métabolisme des neurotransmetteurs). Tout dysfonctionnement mitochondrial (DM) peut donc avoir des effets délétères pouvant notamment induire des maladies neurologiques (ou y contribuer) ou pouvant aggraver les conséquences et la morbidité d'autres anomalies (autisme ou schizophrénie par exemple)[57].
En outre, au cours de la vie, l’accumulation de dommages mitochondriaux contribue au vieillissement et aux maladies neurodégénératives[57].
- myopathies ;
- maladies neurodégénératives ;
- ataxie de Friedreich : maladie touchant la frataxine (Protéine mitochondriale contribuant à l'imperméabilité des membranes au fer) ;
- neuropathie optique de Leber ;
- atrophie optique type I (maladie de Kjer) ;
- hypotonie musculaire ;
- leucodystrophie ;
- microcéphalie ;
- paraplégie spastique ;
- Leucoencéphalophatie
- Ataxie
- MERRF, MELAS, Kearns-Sayre ;
- Encéphalophatie, Leigh ;
- hypothyroïdie ;
- insuffisance hépatocellulaire, Alpers ;
- neuropathie périphérique ;
- retard de croissance ;
- diabète, insuffisance pancréatique ;
- cardiomyopathie hypertrophique ;
- stéatose hépatique ;
- paragangliomes ;
- surdité neurosensorielle ;
- déficit en hormone de croissance ;
- anémie sidéroblastique, syndrome de Pearson ;
- trichothiodystrophie[58].
- Maladie de charcot-Marie tooth
- Chondrosarcome
- Cancer colorectal
- Glioblastome
- Phéochromocytomes
- Cancers du rein
- Syndrome de Cowden
- Leucémie myéloide sévére[59]
- Autisme ? (ou forme plus "sévère" de l'autisme dans certains cas d'anomalie de l'ADN mitochondrial)[57]
- Schizophrénie ? (ou forme plus "sévère" de schizophrénie dans certains cas d'anomalie de l'ADN mitochondrial)[57].
Historique

En 1857, Kölliker décrit les aspects de la mitochondrie dans le muscle. En 1890, Altmann décrit une technique de coloration des mitochondries qu'il appelle bioblastes et postule leur autonomie métabolique et génétique. Mais c’est le microbiologiste et endocrinologue Carl Benda qui, reprenant ces observations sur des préparations colorées au cristal violet, propose en 1898 d’appeler ces structures mitochondria.
En 1937, un scientifique allemand, Hans Adolf Krebs, élabore un modèle de voie métabolique connu sous le nom de cycle de Krebs qui se déroule, chez les eucaryotes, dans les mitochondries. En 1940-1943, Claude isole les mitochondries dans des cellules du foie. En 1948-1950, Kennedy et Lehninger (en) montrent que le cycle de Krebs, la β-oxydation et la phosphorylation oxydative ont tous lieu dans les mitochondries. En 1978, Peter Mitchell obtient le prix Nobel pour sa théorie chimiosmotique. En 1981, Anderson et son équipe découvrent la structure génétique de l’ADN mitochondrial humain. Finalement, Boyer et Walker obtiennent également le prix Nobel pour leurs études sur la structure et le fonctionnement de l'ATP synthase.
En 2016, on ne connaissait qu'un seul eucaryote ayant perdu toutes ses mitochondries : Monocercomonoides (en) sp. PA203[60].
Mobilité des mitochondries dans les cellules
Le réseau de microtubules permet aux mitochondries de se déplacer rapidement là où la cellule a besoin d'énergie. Dans la cellule du tissu musculaire strié squelettique, elles seront disposées près du matériel contractile.
Les mitochondries sont cependant immobiles dans le spermatozoïde car elles sont disposées autour de l'axonème (structure qui compose le flagelle).
Elles le sont également dans les cardiomyocytes et lorsque la cellule est en mitose.
Mobilité des mitochondries en dehors des cellules
En 2020, Alain Thierry, directeur de Recherche Inserm à l’Institut de recherche en cancérologie de Montpellier publie dans la revue scientifique FASEBW[61] les résultats de ses recherches concernant la découverte de mitochondries extra-cellulaires. Pendant sept ans, il a analysé avec son équipe une centaine d'échantillons de plasma sanguin, dans laquelle ils ont détecté des mitochondries libres. Cette découverte permet d'envisager de nouvelles pistes thérapeutiques concernant les diagnostics et les réponses immunitaires du corps. Une nouvelle hypothèse sur la communication entre les cellules est aussi envisagée grâce à cette découverte[62].
Notes et références
- (en) Katrin Henze1 et William Martin, « Evolutionary biology: essence of mitochondria », Nature, vol. 426, no 6963, , p. 127-128 (PMID 14614484, DOI 10.1038/426127a, lire en ligne).
- (en) Lyle Wiemerslage et Daewoo Leeb, « Quantification of mitochondrial morphology in neurites of dopaminergic neurons using multiple parameters », Journal of Neuroscience Methods, vol. 262, , p. 56-65 (PMID 26777473, DOI 10.1016/j.jneumeth.2016.01.008, lire en ligne).
- (en) Heidi M. McBride, Margaret Neuspiel et Sylwia Wasiak, « Mitochondria: more than just a powerhouse », Current biology: CB, vol. 16, no 14, , R551–560 (ISSN 0960-9822, PMID 16860735, DOI 10.1016/j.cub.2006.06.054, lire en ligne, consulté le ).
- (en) Ann Gardner et Richard Boles, « Is a "Mitochondrial Psychiatry" in the Future? A Review », Current Psychiatry Reviews, vol. 1, no 3, , p. 255–271 (DOI 10.2174/157340005774575064, lire en ligne, consulté le ).
- (en) Edward J. Lesnefsky, Shadi Moghaddas, Bernard Tandler et Janos Kerner, « Mitochondrial Dysfunction in Cardiac Disease: Ischemia–Reperfusion, Aging, and Heart Failure », Journal of Molecular and Cellular Cardiology, vol. 33, no 6, , p. 1065-1089 (PMID 11444914, DOI 10.1006/jmcc.2001.1378, lire en ligne, consulté le ).
- (en) Heidi M. McBride, Margaret Neuspiel et Sylwia Wasiak, « Mitochondria: More Than Just a Powerhouse », Current Biology, vol. 16, no 14, , R551-R560 (PMID 16860735, DOI 10.1016/j.cub.2006.06.054, lire en ligne).
- (en) Teresa Valero, « Editorial (Thematic Issue: Mitochondrial Biogenesis: Pharmacological Approaches) », Current Pharmaceutical Design, vol. 20, no 35, , p. 5507-5509 (PMID 24606795, DOI 10.2174/138161282035140911142118, lire en ligne).
- (en) Fabian Sanchis-Gomar, Jose Luis Garcia-Gimenez, Mari Carmen Gomez-Cabrera et Federico V. Pallardo, « Mitochondrial Biogenesis in Health and Disease. Molecular and Therapeutic Approaches », Current Pharmaceutical Design, vol. 20, no 35, , p. 5619-5633 (PMID 24606801, DOI 10.2174/1381612820666140306095106, lire en ligne).
- (en) Ann Gardner et Richard G. Boles, « Is a "Mitochondrial Psychiatry" in the Future? A Review », Current Psychiatry Reviews, vol. 1, no 3, , p. 255-271 (DOI 10.2174/157340005774575064, lire en ligne).
- (en) Gerald W. Dorn II, Rick B. Vega et Daniel P. Kelly, « Mitochondrial biogenesis and dynamics in the developing and diseased heart », Genes & Development, vol. 29, no 19, , p. 1981-1991 (PMID 26443844, PMCID 4604339, DOI 10.1101/gad.269894.115, lire en ligne).
- (en) Steven W. Taylor, Eoin Fahy, Bing Zhang, Gary M. Glenn, Dale E. Warnock, Sandra Wiley, Anne N. Murphy, Sara P. Gaucher, Roderick A. Capaldi, Bradford W. Gibson et Soumitra S. Ghosh, « Characterization of the human heart mitochondrial proteome », Nature Biotechnology, vol. 21, no 3, , p. 281-286 (PMID 12592411, DOI 10.1038/nbt793, lire en ligne).
- (en) Jun Zhang, Xiaohai Li, Michael Mueller, Yueju Wang, Chenggong Zong, Ning Deng, Thomas M. Vondriska, David A. Liem, Jeong-In Yang, Paavo Korge, Henry Honda, James N. Weiss, Rolf Apweiler and Peipei Ping, « Systematic characterization of the murine mitochondrial proteome using functionally validated cardiac mitochondria », Proteomics, vol. 8, no 8, , p. 1564-1575 (PMID 18348319, PMCID 2799225, DOI 10.1002/pmic.200700851, lire en ligne).
- (en) Jun Zhang, David A. Liem, Michael Mueller, Yueju Wang, Chenggong Zong, Ning Deng, Thomas M. Vondriska, Paavo Korge, Oliver Drews, W. Robb MacLellan, Henry Honda, James N. Weiss, Rolf Apweiler et Peipei Ping, « Altered Proteome Biology of Cardiac Mitochondria Under Stress Conditions », Journal of Proteome, vol. 7, no 6, , p. 2204-2214 (PMID 18484766, PMCID 3805274, DOI 10.1021/pr070371f, lire en ligne).
- (en) G. E. Andersson, Olof Karlberg, Björn Canbäck et Charles G. Kurland, « On the origin of mitochondria: a genomics perspective », Philosophical Transactions B, vol. 358, no 1429, , p. 165-177 (PMID 12594925, PMCID 1693097, DOI 10.1098/rstb.2002.1193, lire en ligne).
- Nedjma Ameziane, Marc Bogard et Jérôme Lamoril, Principes de biologie moléculaire en biologie clinique, Elsevier Masson, (lire en ligne), p. 68.
- Pierre Cau, Raymond Seïte et Andrée Robaglia-Schlupp, Cours de Biologie cellulaire, Ellipses, .
- (en) Johannes M. Herrmann et Walter Neupert, « Protein transport into mitochondria », Current Opinion in Microbiology, vol. 3, no 2, , p. 210-214 (PMID 10744987, DOI 10.1016/S1369-5274(00)00077-1, lire en ligne).
- (en) J. E. Chipuk, L. Bouchier-Hayes et D. R. Green, « Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario », Cell Death and Differentiation, vol. 13, no 8, , p. 1396-1402 (PMID 16710362, DOI 10.1038/sj.cdd.4401963, lire en ligne).
- (en) Teruo Hayashi, Rosario Rizzuto, Gyorgy Hajnoczky et Tsung-Ping Su, « MAM: more than just a housekeeper », Trends in Cell Biology, vol. 19, no 2, , p. 81-88 (PMID 19144519, PMCID 2750097, DOI 10.1016/j.tcb.2008.12.002, lire en ligne).
- Cours de Biologie Cellulaire, Pr Stéphane Delbecq, UFR médecine, Montpellier.
- Pierre Cau et Raymond Seïte, Cours de biologie cellulaire, Ellipses, , 560 p. (ISBN 978-2-7298-1138-9, lire en ligne).
- Administrator, « Facbio.com - Structure », sur www.facbio.com (consulté le ).
- « Index of /media/paces/Grenoble_1112 », sur unf3s.cerimes.fr (consulté le ).
- (en) Jiamei Shen, Tingting Du, Xue Wang et Chunli Duan, « α-Synuclein amino terminus regulates mitochondrial membrane permeability », Brain Research, vol. 1591, , p. 14–26 (ISSN 1872-6240, PMID 25446002, DOI 10.1016/j.brainres.2014.09.046, lire en ligne, consulté le ).
- (en) Divya Padmaraj, Rohit Pande, John H. Miller et Jarek Wosik, « Mitochondrial membrane studies using impedance spectroscopy with parallel pH monitoring », PloS One, vol. 9, no 7, , e101793 (ISSN 1932-6203, PMID 25010497, PMCID PMC4091947, DOI 10.1371/journal.pone.0101793, lire en ligne, consulté le ).
- (en) Jeanie B McMillin et William Dowhan, « Cardiolipin and apoptosis », Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids, vol. 1585, nos 2-3, , p. 97-107 (PMID 12531542, DOI 10.1016/S1388-1981(02)00329-3, lire en ligne).
- Abraham L. Kierszenbaum, Histologie et biologie cellulaire : Une introduction à l'anatomie pathologique, De Boeck Supérieur, , 619 p. (ISBN 978-2-8041-4910-9, lire en ligne).
- Daniel Boujard, Bruno Anselme, Christophe Cullin et Céline Raguenes-Nicol, Biologie cellulaire et moléculaire : Tout le cours en fiches 2e édition : 200 fiches de cours, 400 schémas, 160 QCM et site compagnon, Dunod, , 520 p. (ISBN 978-2-10-072892-3, lire en ligne).
- Gerald Karp, Janet Isawa et Wallace Marshall, Biologie cellulaire et moléculaire, De Boeck Supérieur, (lire en ligne), p. 169-170.
- (en) S. Anderson, A. T. Bankier, B. G. Barrell, M. H. L. de Bruijn, A. R. Coulson, J. Drouin, I. C. Eperon, D. P. Nierlich, B. A. Roe, F. Sanger, P. H. Schreier, A. J. H. Smith, R. Staden et I. G. Young, « Sequence and organization of the human mitochondrial genome », Nature, vol. 290, no 5806, , p. 457-465 (PMID 7219534, DOI 10.1038/290457a0, Bibcode 1981Natur.290..457A, lire en ligne).
- (en) Nico M. van Straalen, Dick Roelofs, An Introduction to Ecological Genomics, OUP Oxford, (lire en ligne), p. 61.
- Elucidating the metabolism of members of the Asgard archaea to help updating models on the origin of the eukaryotic cell
- (en) S.G. Andersson, A. Zomorodipour, J.O. Andersson, T. Sicheritz-Ponten, U.C. Alsmark, R.M. Podowski, A.K. Naslund, A.S. Eriksson, H.H. Winkler & C.G. Kurland, « The genome sequence of Rickettsia prowazekii and the origin of mitochondria », Nature, vol. 396, no 6707, , p. 133–140 (PMID 9823893, DOI 10.1038/24094).
- (en) M.W. Gray, G. Burger & B.F. Lang, « The origin and early evolution of mitochondria », Genome Biology, vol. 2, no 6, , reviews1018.1–1018.5 (PMID 11423013, DOI 10.1186/gb-2001-2-6-reviews1018).
- (en) C.G. Kurland & S.G. Andersson, « Origin and evolution of the mitochondrial proteome », Microbiology and Molecular Biology Reviews, vol. 64, no 4, , p. 786–820 (PMID 11104819).
- (en) Y. Tourmen, O. Baris, P. Dessen, C. Jacques, Y. Malthiery, & P. Reynier, « Structure and chromosomal distribution of human mitochondrial pseudogenes », Genomics, vol. 80, no 1, , p. 71–77 (PMID 12079285, DOI 10.1006/geno.2002.6798).
- (en) Woischnik, M. & C.T. Moraes, « Pattern of organization of human mitochondrial pseudogenes in the nuclear genome », Genome Research, vol. 12, no 6, , p. 885–893 (PMID 12045142, DOI 10.1101/gr.227202).
- M. N. C., « Mitochondries héritées du père », Pour la science, no 496, , p. 16.
- (en) Shiyu Luo, C. Alexander Valencia, Jinglan Zhang, Ni-Chung Lee, Jesse Slone et al., « Biparental Inheritance of Mitochondrial DNA in Humans », PNAS, vol. 115, no 51, , p. 13039-13044 (DOI 10.1073/pnas.1810946115).
- (en) Mensur Dlakić et Arcady Mushegian, « Prp8, the pivotal protein of the spliceosomal catalytic center, evolved from a retroelement-encoded reverse transcriptase », RNA, vol. 17, , p. 799-808 (ISSN 1355-8382 et 1469-9001, PMID 21441348, PMCID 3078730, DOI 10.1261/rna.2396011, lire en ligne, consulté le ).
- (en) Rusch, S.L. & D.A. Kendall, « Protein transport via amino-terminal targeting sequences: common themes in diverse systems », Molecular Membrane Biology, vol. 12, no 4, , p. 295–307 (PMID 8747274, DOI 10.3109/09687689509072431).
- (en) G. Schatz & B. Dobberstein, « Common principles of protein translocation across membranes », Science, vol. 271, no 5255, , p. 1519–1526 (PMID 8599107, DOI 10.1126/science.271.5255.1519).
- (en) M.F. Lopez, B.S. Kristal, E. Chernokalskaya, A. Lazarev, A.I. Shestopalov, A. Bogdanova, & M. Robinson, « High-throughput profiling of the mitochondrial proteome using affinity fractionation and automation », Electrophoresis, vol. 21, no 16, , p. 3427–3440 (PMID 11079563).
- (en) C. Andreoli, H. Prokisch, K. Hortnagel, J.C. Mueller, M. Munsterkotter, C. Scharfe, & T. Meitinger, « MitoP2, an integrated database on mitochondrial proteins in yeast and man », Nucleic Acids Research, vol. 32, no Database issue, , D459–D462 (PMID 14681457, DOI 10.1093/nar/gkh137).
- (en) D. Cotter, P. Guda, E. Fahy, & S. Subramaniam, « MitoProteome: mitochondrial protein sequence database and annotation system », Nucleic Acids Research, vol. 32, no Database issue, , D463–D467 (PMID 14681458, DOI 10.1093/nar/gkh048).
- (en) P. R. Rich, « The molecular machinery of Keilin's respiratory chain », Biochemical Society Transactions, vol. 31, no Pt 6, , p. 1095-1105 (PMID 14641005, DOI 10.1042/BST0311095, lire en ligne).
- Emily E. Noble, Ted M. Hsu et Scott E. Kanoski, « Gut to Brain Dysbiosis: Mechanisms Linking Western Diet Consumption, the Microbiome, and Cognitive Impairment », Frontiers in Behavioral Neuroscience, vol. 11, (ISSN 1662-5153, DOI 10.3389/fnbeh.2017.00009, lire en ligne, consulté le )
- Joseph C. LaManna, Nicolas Salem, Michelle Puchowicz et Bernadette Erokwu, « Ketones Suppress Brain Glucose Consumption », dans Advances in Experimental Medicine and Biology, Springer US (ISBN 9780387859972, lire en ligne), p. 301–306
- (en) Dominique Chretien, Paule Benit, Hyung-Ho Ha et Susanne Keipert, « Mitochondria Are Physiologically Maintained At Close To 50 C », bioRxiv, , p. 133223 (DOI 10.1101/133223, lire en ligne, consulté le ).
- (en) Dominique Chrétien, Paule Bénit, Hyung-Ho Ha et Susanne Keipert, « Mitochondria are physiologically maintained at close to 50 °C », PLOS Biology, vol. 16, no 1, , e2003992 (ISSN 1545-7885, PMID 29370167, PMCID PMC5784887, DOI 10.1371/journal.pbio.2003992, lire en ligne, consulté le ).
- « Les mitochondries travaillent dans la cellule à près de 50 °C », sur le site du CNRS.
- « Au cœur de nos cellules, la température n’est pas de 37 °C », sur Le Figaro, .
- (en) Guillaume Baffou, Hervé Rigneault, Didier Marguet et Ludovic Jullien, « A critique of methods for temperature imaging in single cells », Nature Methods, vol. 11, no 9, , p. 899–901 (ISSN 1548-7091 et 1548-7105, DOI 10.1038/nmeth.3073, lire en ligne, consulté le ).
- (en) Dominique Chrétien, Paule Bénit, Hyung-Ho Ha et Susanne Keipert, « Mitochondria are physiologically maintained at close to 50 °C », PLOS Biology, vol. 16, no 1, (ISSN 1545-7885, DOI 10.1371/annotation/51c92619-8dc5-43a1-8117-05c19db159f5, lire en ligne, consulté le ).
- « Nos mitochondries seraient beaucoup plus chaudes qu'on ne le croyait », sur maxisciences.com, .
- Rose, S., Wong, S., & Giulivi, C. (2016). Mitochondrial DNA Damage in Autism. In Biochemistry of Oxidative Stress (p. 327-343). Springer, Cham (résumé).
- S.Loublier et al., Les maladies mitochondriales : une médecine à part ? 2009 DOI 10.1016/j.immbio.2009.08.002
- P.Bénit et al., Pathologies liées aux déficits du cycle de Krebs. Revue Francophone des laboratoires No 501 2018
- (en) Anna Karnkowska, Vojtěch Vacek, Zuzana Zubáčová, Sebastian C. Treitli, Romana Petrželková, Laura Eme, Lukáš Novák, Vojtěch Žárský, Lael D. Barlow, Emily K. Herman, Petr Soukal, Miluše Hroudová, Pavel Doležal, Courtney W. Stairs, Andrew J. Roger, Marek Eliáš, Joel B. Dacks, Čestmír Vlček et Vladimír Hampl, « A Eukaryote without a Mitochondrial Organelle », Current Biology, vol. 26, no 10, , p. 1274-1284 (PMID 27185558, DOI 10.1016/j.cub.2016.03.053, lire en ligne).
- «Blood contains circulating cell‐free respiratory competent mitochondria», The FASEB Journal, 19 janvier 2020
- «La découverte d'un nouveau composant du sang», Le Journal des Sciences, France Culture, 22 janvier 2020
Voir aussi
Articles connexes
Liens externes
- «Mitochondrie, de l’énergie plein la cellule», La Méthode scientifique, France Culture, 24 juin 2019
- «La découverte d'un nouveau composant du sang», Le Journal des sciences, La Méthode scientifique, France Culture, 22 janvier 2019
- [vidéo] (27 minutes) : La génétique de la mitochondrie chez les paramécies
- [vidéo] (4 minutes) : Cours : La mitochondrie
- Les mitochondries - eBiologie.fr