Substitution nucléophile d'acyle
La substitution nucléophile d'acyle est un type de réaction de substitution entre un nucléophile et un composé porteur d'un groupe acyle. Dans ce type de réaction, le nucléophile (un alcool, une amine, ou un énolate par exemple) va se substituer au nucléofuge (ou groupe partant) du dérivé acyle (halogénure d'acyle, anhydride, ou ester). Comme les dérivés acyle réagissent avec un grand nombre de nucléophiles, et comme le produit final dépend du type particulier de dérivé d'acyle impliqué, les réactions de substitution nucléophile d'acyle peuvent être utilisées pour synthétiser une large gamme de produits.
Mécanisme réactionnel
Les composés carbonylés réagissent avec les nucléophiles par un mécanisme deux étapes. La première est une étape de type addition : le nucléophile attaque le carbone du carbonyle, formant un intermédiaire tétraédrique (en). Cette réaction peut être accélérée en conditions acides, ce qui rend le carbonyle plus électrophile, ou basiques, ce qui rend le nucléophile plus anionique et donc plus réactif. L'intermédiaire tétraédrique peut lui-même être un alcool ou un alcoolate, en fonction du pH du milieu réactionnel.
La seconde étape est du type élimination : l'intermédiaire tétraédrique contient un substituant attaché au carbone central qui peut agir comme nucléofuge. Ainsi, aussitôt formé, l'intermédiaire tétraédrique se décompose, recréant une liaison double C=O et en éjectant le nucléofuge. Ces deux étapes sont réversibles, faisant des réactions de substitution nucléophile d'acyle des réactions d'équilibre[1]. Comme cet équilibre favorise le produit contenant le meilleur nucléophile, le nucléofuge doit donc être comparativement un nucléophile médiocre, afin que la réaction ait un intérêt pratique.
Conditions acides

En conditions acides, le groupe carbonyle du composé acylé (1) est protoné, ce qui l'active en vue d'une attaque nucléophile. Le carbonyle protoné (2) est ensuite attaqué par la nucléophile (H−Z) pour former l'intermédiaire tétraédrique (3). Un transfert de proton depuis le nucléophile (Z) vers le nucléofuge (X) donne l'intermédiaire 4, qui se décompose en éjectant le nucléofuge protoné (H−X), formant un composé carbonylé (5). La perte d'un proton donne le produit de substitution (6). Comme la dernière étape implique la perte d'un proton, les réactions de substitution nucléophile d'acyle sont considérées comme catalysées par les acides. À noter aussi que sous conditions acides, le nucléophile sera typiquement sous forme protonée (c'est-à-dire H−Z au lieu de Z−).
Conditions basiques
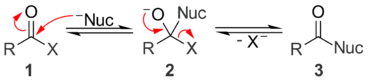
En conditions basiques, le nucléophile (Nuc) attaque le groupe carbonyle du composé acylé (1) pour donner un intermédiaire tétraédrique alcoolate (2). Cet intermédiaire se décompose en expulsant le nucléofuge (X), formant le produit de substitution (3). Si cette action des bases est catalytique dans certains cas de substitution nucléophile d'acyle, elle ne l'est plus si le nucléofuge est une base plus faible que le nucléophile. Contrairement aux réactions sous catalyse acide, le nucléophile et les nucléofuge sont sous forme anionique en conditions basiques.

Ce mécanisme est confirmé par des expériences avec marquage isotopique. Lorsque le propanoate d'éthyle dont le groupe éthoxy est marqué à l'oxygène 18 est traité par l'hydroxyde de sodium (NaOH), l'oxygène 18 est complètement absent de l'acide propanoïque formé, et est exclusivement trouvé dans l'éthanol[2].
Réactivité
Il existe cinq principaux dérivés acyle : les halogénures d'acyle, qui sont les plus réactifs envers les nucléophiles, suivis par les anhydrides, les esters, et les amides. Les ions carboxylates sont quasiment non-réactifs par substitution nucléophile d'acyle puisqu'ils ne possèdent pas de nucléofuge. Cette différence de réactivité entre ces cinq familles de classes de composés couvre une large gamme ; par exemple, le taux de réactivé relatif entre les chlorures d'acyle et les amides diffère d'un facteur dix13[3].
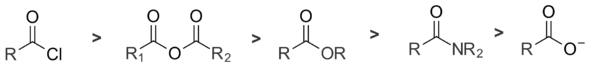
Un facteur majeur déterminant la réactivité des dérivé acyle est la capacité du nucléofuge à partir, ce qui est lié à son acidité / sa basicité. Une espèce avec un fort acide conjugué (par exemple l'acide chlorhydrique) sera un meilleur nucléofuge qu'une espèce avec un acide conjugué faible (par exemple l'acide acétique) ; ainsi, les ions chlorures sont des meilleurs nucléofuges que les ions acétate. Les bases faibles sont donc de meilleurs groupes partants que les bases fortes et la réactivité des dérivés acyle envers les nucléophiles diminue quand la basicité du nucléofuge augmente[4].
| Nom du composé | Structure | Nucléofuge | pKa du couple nucléofuge |
|---|---|---|---|
| chlorure d'acétyle | 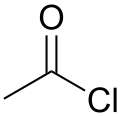 |
−7 | |
| anhydride acétique | 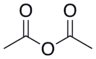 |
 |
4.76 |
| acétate d'éthyle | 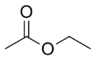 |
15.9 | |
| acétamide | 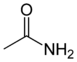 |
38 | |
| anion acétate | |
N/a | N/a |
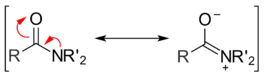
Un autre facteur qui joue un rôle dans la réactivité des composés acyle est la mésomérie. Les amides présentent deux formes mésomères, chacune contribuant à la forme réelle de la molécule, si bien que la liaison amide entre l'atome de carbone du groupe carbonyle et l'atome d'azote possède un caractère important de liaison double. La barrière énergétique à la rotation autour de la liaison amide est de 75–85 kJ/mol, bien plus grande que les valeurs observées pour les liaisons simples. Par exemple, pour la liaison simple C-C de l'éthane, la barrière énergétique est seulement de 12 kJ/mol[3]. Lorsqu'un nucléophile attaque un amide formant l'intermédiaire tétraédrique, cet effet mésomère énergétiquement favorable disparaît. Ceci aide à comprendre pourquoi les amides sont parmi les mois réactifs des dérivés acyle[4].
Les esters possèdent le même genre d'effet mésomère, mais bien moins fort que dans le cas des amides, ainsi la formation de l'intermédiaire tétraédrique et la perte de mésomérie est moins énergétiquement défavorable. Dans les anhydrides, cet effet est encore moindre, car la mésomérie est partagée entre les deux groupes carbonyle, et elle est quasiment inexistante pour les halogénures d'acyle. Ceci permet une fois de plus de comprendre l'ordre de réactivité entre les dérivés acyle[4].
Réactions des dérivés acyle
La plupart des substitutions nucléophiles d'acyle sont des conversions d'un dérivé acyle en un autre. En général, on doit convertir un dérivé acyle en un moins réactif pour que la réaction soit efficace, le produit devant être plus stable que le composé de départ. On peut ainsi facilement convertir un chlorure d'acyle en ester, mais la réaction inverse est en pratique quasiment impossible.
Les substitutions nucléophiles d'acyle peuvent aussi convertir un dérivé d'acyle en une autre espèce. Par exemple, les amides et les acides carboxyliques peuvent réagir avec les réactifs de Grignard pour produire des cétones. La suite du paragraphe présente un aperçu des réactions possibles pour chacune des classes de dérivés d'acyle.
Halogénures d'acyle
Les halogénures d'acyle sont les plus réactifs des dérivé acyle et peuvent être facilement convertis en n'importe quel autre. Ils peuvent par exemple réagir avec les acides carboxyliques pour former des anhydrides ; si les substituants de l'acide carboxylique et de l'halogénure sont différents, le produit final sera un anhydride mixte.

La figure ci-dessus présente la réaction du chlorure de benzoyle avec l'acide acétique. Dans un premier temps, l'acide va attaquer le chlorure de benzoyle (1) pour former un intermédiaire tétraédrique (2). Cet intermédiaire va se réorganiser en expulsant un ion chlorure (nucléofuge), et formant un ion oxonium (3). Cet ion va subir une déprotonation et donner l'anhydride mixte (4) et un équivalent d'acide chlorhydrique.
Les alcools et les amines réagissent avec les halogénures d'acyle pour donner des esters et des amides, respectivement, dans une réaction connue sous le nom de réaction de Schotten-Baumann[5].
Les halogénures d'acyle s'hydrolysent en présence d'eau pour donner les acides carboxyliques équivalents, mais cette réaction est rarement utile puisqu'on utilise en général les acides carboxyliques pour produire ces halogénures d'acyle (notamment par réaction avec des agents d'halogénation comme le chlorure de thionyle).
La plupart des réactions avec les halogénures d'acyle sont effectuées en présence d'une base non-nucléophile, comme la pyridine, pour neutraliser l'acide halogénohydrique produit en même temps que le produit désiré.
Les halogénures d'acyle réagissent avec les carbones nucléophiles, comme les réactifs de Grignard et les énolates, donnant en général des mélanges de produits. En effet, si le carbone nucléophile réagit d'abord avec l'halogénure d'acyle pour donner une cétone, cette dernière peut ensuite aussi subir une attaque nucléophile pour former un alcool tertiaire.

Dans l'exemple ci-dessus, le chlorure de benzoyle (1) est traité par deux équivalents d'un réactif de Grignard, ici le bromure de méthylmagnésium (MeMgBr), formant le 2-phénylpropan-2-ol (3) avec un très bon rendement (92 %). Même si l'acétophénone (2) est formée comme intermédiaire de réaction, il est impossible de l'isoler car elle réagit rapidement avec le second intermédiaire de MeMgBr après sa formation[6].
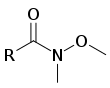
Contrairement aux autres carbones nucléophiles, les dialkylcuprates lithiés – souvent appelés réactifs de Gilman – peuvent réagir une seule fois avec les halogénures d'acyle en produisant des cétones. Cette réaction n'est cependant pas une substitution nucléophile d'acyle, et on pense que le mécanisme de cette réaction est radicalaire[2]. La synthèse de cétone de Weinreb (en) peut aussi être utilisée pour convertir les halogénures d'acyle en cétones. Dans cette réaction, l'halogénure d'acyle est d'abord converti en un N–méthoxy–N–méthylamide, appelé amide de Weinreb. Lorsqu'un carbone nucléophile (réactif de Grignard ou organolithien) réagit avec cette amide de Weinreb, le métal est chélaté par les oxygènes du carbonyle et du groupe N–méthoxy, empêchant de nouvelle additions nucléophiles[7].
Dans l'acylation de Friedel-Crafts, les halogénures d'acyle agissent comme électrophiles dans des réactions de substitution électrophile aromatique. Un acide de Lewis, tel que le chlorure de zinc (ZnCl2), le chlorure de fer(III) (FeCl3), ou le chlorure d'aluminium (AlCl3), se coordonne avec l'halogène de l'halogénure d'acyle, activant le composé pour une attaque nucléophile. Pour les cycles aromatique spécialement riches en électrons, la réaction se produit sans l'ajout d'acide de Lewis[8].
Thioesters
Les thioesters réagissent de façon similaire aux chlorures d'acyle, mais de façon plus douce.
Anhydrides
La chimie des halogénures d'acyle et des anhydrides est similaire. Si les anhydrides ne peuvent être convertis en halogénure d'aycle, ils peuvent être convertis en chacun des autres dérivés acyle. Les anhydrides peuvent aussi réagir par des réactions similaires à la réaction de Schotten-Baumann pour produire des esters et des amides à partir d'alcools et d'amines, et l'eau peut les hydrolyser en leurs acides correspondants. Comme les halogénures d'acyle, les anhydrides peuvent réagir avec les carbones nucléophiles pour produire des cétones et/ou des alcools tertiaires, et peuvent participer aux acylations de Friedel–Crafts comme aux synthèses de cétone de Weinreb[8]. Cependant, contrairement aux halogénures d'acyle, les anhydrides ne réagissent pas avec les réactifs de Gilman[2].
La réactivité des anhydrides peut être accrue en ajoutant une quantité catalytique de N,N-diméthylaminopyridine (DMAP). La pyridine peut aussi être utilisée dans ce but, et agit de façon similaire[5].

Dans un premier temps, la DMAP (2) attaque l'anhydride (1) pour former un intermédiaire tétraédrique, qui se réorganise en éjectant un ion carboxylate et donnant un amide (3). Cet amide intermédiaire est plus actif pour les attaques nucléophiles que l'anhydride initial car la diméthylaminopyridine est un meilleur nucléofuge que l'ion carboxylate. Dans une étape suivante, un nucléophile (Nuc) attaque 3 pour donner un autre intermédiaire tétraédrique qui va se réorganiser pour donner le produit final (4), en éjectant le groupe pyridine, ce qui lui rend au passage son caractère aromatique, un élément qui favorise grandement la réaction et explique pourquoi il est un meilleur nucléofuge que l'ion carboxylate.
Esters
Les esters sont moins réactifs que les halogénures d'acyle et les anhydrides. Comme les dérivés acyle plus réactifs, ils peuvent réagir avec l'ammoniac et les amines primaires et secondaires pour donner des amides, mais ce type de réaction n'est guère utilisé car les réactions avec les halogénure d'acyles donnent de meilleurs rendements. Les esters peuvent être transformés en d'autre esters, un processus appelé transestérification. La transestérification peut être catalysée en milieu acide ou basique, et implique la réaction entre un ester et un alcool. Malheureusement, comme le nucléofuge est un alcool, la réaction directe et inverse se déroulent en même temps, à des vitesses similaires. En utilisant l'alcool réactif en large excès ou en extrayant l'alcool partant (par exemple par distillation) favorisera la réaction dans le sens direct, selon le principe de Le Chatelier[9].
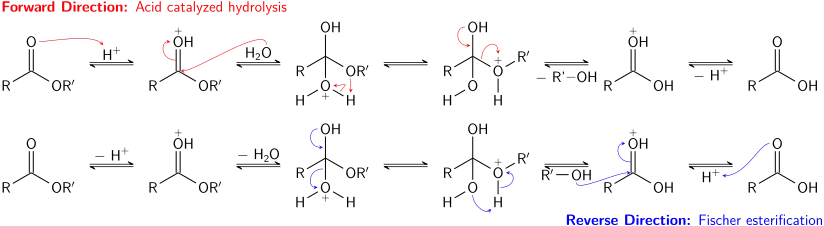
L'hydrolyse acide des esters est une réaction d'équilibre, essentiellement la réaction inverse de l'estérification de Fischer. Comme les alcools (nucléofuge) et l'eau (nucléophile) ont un pKa similaire, les réactions directes et inverses se retrouvent en compétition. Comme dans le cas de la trans-estérification, utiliser un réactif (l'eau) en large excès, ou extraire le produit (l'alcool) au fur et à mesure de sa production permet de favoriser la réaction directe.
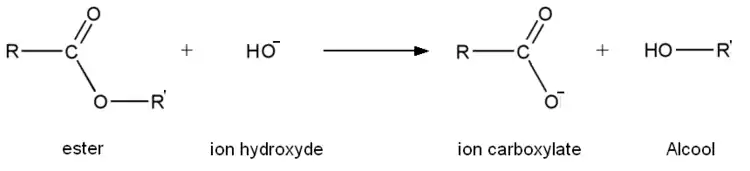
L'hydrolyse basique des esters, connue sous le nom de saponification n'est en revanche pas une réaction d'équilibre ; c'est une réaction totale qui consomme un équivalent de base dans la réaction, et produit un équivalent d'alcool et un équivalent d'ion carboxylate. La saponification des esters d'acides gras est un procédé industriel important, utilisé dans la production de savon[9].

Les esters peuvent subir un certain nombre de réactions avec les carbones nucléophiles. Comme les halogénures d'acyle et les anhydrides, ils réagissent avec les réactifs de Grignard en excès pour donner des alcools tertiaires. Ils peuvent également réagir avec les énolates. Dans la condensation de Claisen, un énolate d'un ester (1) va attaquer le carbone du groupe carboxyle d'un autre ester (2) pour former un intermédiaire tétraédrique (3). Cet intermédiaire se réorganise en expulsant un groupe alcoolate (R'O−), produisant un β-cétoester (4).
Les condensations de Claisen croisées, c'est-à-dire dans lesquelles l'énolate et le nucléophiles sont deux esters différents sont possibles, mais produisent des mélanges de composés qu'il faut ensuite séparer. Une condensation de Claisen intramoléculaire (d'une molécule avec deux fonctions ester) est appelée condensation de Dieckmann ou cyclisation de Dieckmann, car elle est utilisée pour former des cycles. Les esters peuvent aussi subir des condensations avec des cétones et des aldéhydes énolisables, pour donner des composés β-dicarbonylés[10]. Un exemple spécifique est le réarrangement de Baker-Venkataraman dans lequel une ortho-acyloxycétone aromatique subit une substitution nucléophile d'acyle intramoléculaire suivi par un rearrangement pour former une β-dicétone aromatique[11]. Le réarrangement de Chan est un autre exemple de réarrangement résultant d'une substitution nucléophile d'acyle intramoléculaire.
Amides
Du fait de leur faible réactivité, les amides ne participent pas à autant de types de substitution nucléophiles que les autres dérivés acyle. Les amides sont stables en présence d'eau, et sont environ cent fois plus stable vis-à-vis de l'hydrolyse que les esters[3]. Les amides peuvent cependant être hydrolysés en acides carboxyliques en présence d'acide ou de base. La stabilité des liaisons amide a des implications biologiques, du fait que les acides aminés formant les protéines sont liés entre eux par des liaisons amide. Les liaisons amide sont suffisamment résistantes à l'hydrolyse pour maintenir la protéine en forme dans des environnements aqueux mais peuvent cependant être brisées si nécessaire[3].
Les amides primaires non-substitués (R-C(=O)NH2) ou momnosubstitués (R-C(=O)NHR') ne réagissent pas favorablement avec les carbones nucléophiles. Les organomagnésiens et les organolithiens réagissent avec eux en tant que base plutôt qu'en tant que nucléophile, et vont simplement les déprotoner. Les amides primaires disubstitués (R-C(=O)NR'R'') n'ont pas ce problème et réagissent avec les carbones nucléophiles pour former des cétones ; les anions amidure (NR2−) sont des bases très fortes et donc de très mauvais nucléofuges, l'attaque nucléophile ne se produira donc qu'une seule fois. Lorsqu'il réagit avec un des carbones nucléophiles, le N,N-diméthylformamide (DMF) peut être utilisé pour introduire un groupe formyle[12].

Dans l'exemple ci-dessus, le phényllithium (1) attaque le groupe carbonyle du DMF (2), formant un intermédiaire tétraédrique (3). Comme l'anion diméthylamidure est un mauvais nucléofuge, l'intermédiaire ne va pas l'expulser, et aucune autre attaque nucléophile ne peut se produire. Sous conditions acides, le groupe alcoolate va se protoner pour former un aminoalcool (4), puis le groupe amine va également se protoner pour former un ion ammonium (5). L'élimination de la diméthylamine, une molécule neutre, et la perte d'un proton, donne le benzaldéhyde (6).
Acides carboxyliques
Les acides carboxyliques ne sont pas spécialement réactifs par substitution nucléophile, mais ils peuvent être convertis en d'autres dérivés acyle.
Convertir un acide carboxylique en amide est possible, mais ce n'est pas une réaction directe et simple. Au lieu de réagir comme nucléophile, une amine réagira comme une base en présence d'un acide carboxylique, produisant un carboxylate d'ammonium. Chauffer le sel ainsi obtenu au-dessus de 100 °C permettra d'éliminer l'eau et de former l'amide. Cette méthode de synthèse des amides est importante dans le monde industriel, comme pour les applications en laboratoire[13].
En présence d'un acide fort catalyseur, les acides carboxyliques peuvent se condenser pour former des anhydrides d'acide. Cette condensation produit cependant de l'eau qui peut réhydrolyser l'anhydride obtenu en l'acide carboxylique initial. Cette réaction est donc une réaction d'équilibre.
Sous catalyse acide, les acides carboxyliques peuvent réagir avec les alcools pour former des esters, par l'estérification de Fischer, qui est également une réaction d'équilibre. On peut aussi utiliser le diazométhane pour convertir un acide en ester. Si cette réaction offre de bon rendements, elle ne permet cependant que de produire des esters de méthyle[13].
Le chlorure de thionyle peut être utilisé pour convertir les acides carboxyliques en leur chlorures d'acyle équivalents. Dans un premier temps, l'acide carboxylique (1) attaque le chlorure de thionyle et provoque le départ d'un ion chlorure. L'ion oxonium (2) ainsi résultant est activé en vue d'une attaque nucléophile, tout en possédant un bon nucléofuge, contrairement à un acide carboxylique normal. Dans l'étape suivante, cet ion est attaqué par un ion chlorure pour donner un intermédiaire tétraédrique (3), un chlorosulfite. Cet intermédiaire se réorganise éjectant un dioxyde de soufre et un ion chlorure, produisant un chlorure d'acyle protoné (4). L'ion chlorure peut retirer ce proton du groupe carbonyle, donnant le chlorure d'acyle final (5), avec production de HCl.

Le chlorure de phosphore(III) (PCl3) et chlorure de phosphore(V) (PCl5) permettent également de convertir un acide carboxylique en chlorure d'acyle, par un mécanisme similaire. Un équivalent de PCl3 peut réagir avec trois équivalents d'acide, produisant un équivalent de H3PO3, ou acide phosphoreux, en plus du chlorure d'acyle désiré. PCl5 réagit avec les acides carboxyliques dans un ratio 1:1, et produit l'oxychlorure de phosphore(V) (POCl3) comme sous-produit.
Les acides carboxyliques peut réagir avec les organomagnésiens ou les organolithiens pour former des cétones. Un premier équivalent de nucléophile agira comme base qui déprotenera l'acide. Un second équivalent attaquera le groupe carbonyle pour former un anion alcoolate géminal, qui, protoné, donnera un hydrate de cétone. Comme la plupart des hydrates de cétone sont instables par rapport à leur cétone correspondante, l'équilibre entre les deux formes est fortement déplacé en faveur des cétones. Par exemple, la constante d'équilibre de la formation de l'hydrate d'acétone à partir de l'acétone est seulement de 0,002[14].
Notes et références
- Wade 2010, p. 996–997.
- John McMurry, Organic Chemistry, Pacifc Grove, CA, Brooks/Cole Publishing Company, , 4e éd. (ISBN 0-534-23832-7), p. 820–821
- Francis A. Carey, Organic Chemistry, New York, McGraw-Hill, , 6e éd. (ISBN 0-07-282837-4), p. 866–868
- Wade 2010, p. 998–999.
- (en) László Kürti et Barbara Czakó, Strategic Applications of Named Reactions in Organic Synthesis : background and detailed mechanisms, Londres, Elsevier Academic Press, , 398 p. (ISBN 0-12-429785-4)
- McMurry 1996, p. 826–827.
- Kürti and Czakó 2005, p. 478.
- Kürti and Czakó 2005, p. 176.
- Wade 2010, p. 1005–1009.
- Carey 2006, p. 919–924.
- Kürti and Czakó 2005, p. 30.
- Alan R. Katritzky, Otto Meth-Cohn et Charles W. Rees, Comprehensive Organic Functional Group Transformations, vol. 3, Oxford, Pergamon Press, , 1re éd. (ISBN 0-08-042324-8), p. 90
- Wade 2010, p. 964–965.
- Wade 2010, p. 838.
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Nucleophilic acyl substitution » (voir la liste des auteurs).
Voir aussi
- Abstraction nucléophile
Liens externes
- Réaction de l'anhydride acétique avec l'acétone in Organic Syntheses Coll. vol. 3, p. 16, vol. 20, p. 6, Article