Corrosion aqueuse
La corrosion désigne l'altération d'un objet manufacturé par l'environnement. Les exemples les plus connus sont les altérations chimiques des métaux dans l'eau — avec ou sans oxygène — telles la rouille du fer et de l'acier, ou la formation de vert-de-gris sur le cuivre et ses alliages (bronze, laiton). Ces altérations chimiques sont regroupées sous le terme de corrosion aqueuse. Elles sont dues à des effets de plusieurs sortes : dissolution des métaux dans l'eau, apparition de piles électrochimiques, existence de gradients de concentration, aération différentielle ou piqûration. Globalement, la corrosion aqueuse est un phénomène dont l'impact économique est très important, nécessitant une grande variété de moyens de protections des métaux.
Corrosion généralisée : la dissolution des métaux
La corrosion des métaux consiste essentiellement en leur oxydation, qui est un « retour à l'état naturel. » Cependant, l'oxydation n'est pas nécessairement la combinaison d'un élément avec de l'oxygène. D'une manière plus générale, il s'agit d'une réaction chimique au cours de laquelle un composé considéré cède des électrons.
Par exemple, le fer s'oxyde en présence de l'oxygène de l'air pour former différents types d'oxydes de Fe(III) plus ou moins hydratés dont notamment l'hématite (Fe2O3) (cas le plus simple de l'oxyde anhydre) comme illustré schématiquement ci-dessous :
- Oxydation du fer (normalisée à 12 électrons) :

- Réduction de l'oxygène (normalisée à 12 électrons) :

- Réaction globale :

L'hématite peut être décrite comme un cristal ionique (Fe3+2, O2−3), ce qui n'est pas tout à fait exact, mais donne une bonne approche du phénomène d'oxydation. Au cours de la réaction, le fer cède des électrons : on dit qu'il est oxydé (son étage d'oxydation augmente de 0 à 3+). L'oxygène capte des électrons : on dit qu'il est réduit (son étage d'oxydation diminue de 0 à 2-).
La corrosion des métaux en milieu aqueux résulte de l'oxydation du métal, mais pas nécessairement par l'oxygène de l'air qui est dissous dans l'eau : l'oxydation peut également se produire avec d'autres espèces chimiques, notamment des protons hydratés: ions H+ ou H3O+.
Il est toutefois important de noter la formation d’autres formes d’oxydes de fer comme la magnétite Fe3O4 de structure spinelle et qui est un oxyde mixte de fer(II) et de fer(III). Ce produit est stable dans l’eau y compris en absence d’oxygène en accord avec le diagramme de Pourbaix.
Lorsque l'on plonge du fer dans une solution acide (pH < 7), le fer se dissout avec un dégagement d'hydrogène. En fait, il s'agit là d'une version accélérée de la corrosion en milieu aqueux en absence d'oxygène:



Les ions Fe3+ passent alors en solution aqueuse et sont solubles en condition acide (très bas pH). Si la valeur du pH augmente, les ions Fe3+ s'hydrolysent et se combinent avec les ions OH− produits par l'autoprotolyse de l'eau et forment des hydroxydes ou des oxy-hydroxides de Fe(III), comme Fe(OH)3 ou FeO(OH).
Cette oxydation par les protons présents dans l'eau est rapide en milieu acide. Elle reste possible, quoique beaucoup plus lente, en solution neutre (pH = 7) mais se ralentit considérablement en conditions très basiques (pH > 10). La raison est que le nombre de protons capables d'accepter des électrons diminue fortement à haut pH et aussi que la solubilité des hydroxydes et des oxydes est très faible en conditions basiques (effet d'ions communs avec les OH−).
La corrosion uniforme de la surface du fer par l'oxygène dissout dans l'eau ou suite à l'oxydation par les protons de l'eau (en absence d'oxygène) conduit à la corrosion généralisée de l'objet ou de la pièce métallique. La présence d'eau à la surface du métal et l'humidité de l'air accélèrent également les processus de corrosion en augmentant la mobilité des ions dissous en phase aqueuse. En milieu très sec, l'oxydation du fer et des aciers est beaucoup plus lente, car le transport des ions est fortement ralenti ou inexistant. L'augmentation de la concentration en sels dissous dans l'eau (électrolytes, p. ex. NaCl) favorise également les processus de corrosion (voir plus loin la corrosion localisée et la corrosion par piqûre). L'eau et l'humidité jouent donc un rôle capital dans les processus de corrosion où la phase aqueuse participe activement aux mécanismes d'oxydation.
L'oxydation des métaux peut cependant aussi se produire directement à l'interface métal/gaz sans devoir passer nécessairement par un processus de dissolution. Ainsi, l'oxygène réagit-il directement avec l'aluminium pour former une couche d'oxyde (alumine, Al2O3). Si cette couche est continue (c.-à-d., ininterrompue, sans fissure, ni perforation), compacte (c.-à-d., dense, très peu poreuse) et adhérente (pas de cloque ou de délamination), elle est très peu perméable à l'oxygène et protège ainsi la pièce de l'oxydation ultérieure. Le transport de l'oxygène gazeux au travers du film d'oxyde est négligeable : pas d'advection ni de diffusion de l'O2 gazeux au sein du film d'Al2O3 recouvrant uniformément la surface de l'aluminium métallique sous-jacent. On dit alors que le métal est passivé. Une pièce passivée continue à s'oxyder, mais à une vitesse extrêmement lente, la couche d'oxyde dite « passive » faisant écran. La pièce est donc considérée comme durablement protégée contre la corrosion.
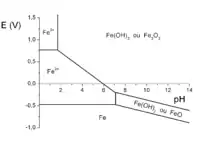
La stabilité du fer dans l'eau dépend :
- du pH, qui détermine la concentration d'ions H3O+ dans l'eau ;
- du potentiel électrique de la pièce en fer par rapport à la solution, qui détermine la capacité des électrons à quitter le fer.
On peut ainsi tracer un diagramme potentiel-pH (Eh, pH), en indiquant les zones de stabilité du fer (Fe), les zones de stabilité de l'ion Fe2+ (ou « ion fer II »), les zones de stabilité de l'ion Fe3+ (ou « ion fer III ») et les zones de passivation. Il s'agit donc d'une sorte de « carte », les zones délimitées par des frontières indiquant les couples de valeurs (Eh, pH) pour lesquelles une espèce est stable. Ce diagramme porte le nom de diagramme de Pourbaix, et peut être tracé pour tous les métaux.
Pour savoir si un matériau est adapté à un milieu, il suffit de regarder le diagramme de Pourbaix de ce matériau. Si le couple (Eh, pH) se situe dans une zone de stabilité, le matériau est protégé contre la corrosion généralisée.
Corrosion galvanique (pile électrochimique)
Généralités
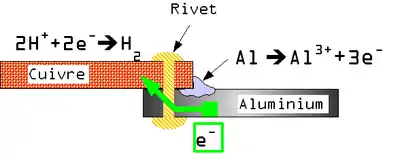
Une pile électrochimique est créée lorsque deux métaux de natures différentes sont mis en contact. Un des métaux s'oxyde et se dissout (anode), tandis que sur l'autre métal a lieu une réduction (cathode), et éventuellement formation d'une couche de produits de réaction (des espèces chimiques de la solution se réduisent et se déposent, notamment dépôt calco-magnésien). On parle de corrosion galvanique. Ce phénomène explique :
- le principe de la « protection cathodique par anode sacrificielle » : on crée une pile électrochimique qui impose un sens de parcours aux électrons pour empêcher la réaction de corrosion ; l'anode se dissout (elle est sacrifiée) et la cathode reste stable, elle est de plus parfois protégée par une couche de produits de réaction ;
- le principe de la « protection cathodique par courant imposé » : à la place de l'anode sacrificielle, on peut imposer le sens de parcours des électrons en établissant une différence de potentiel entre la pièce et le milieu avec un générateur de tension, par exemple alimenté par des panneaux solaires ;
- pourquoi lorsque l'on met deux métaux différents en contact, l'un se corrode très rapidement.
C'est exactement le même type de réactions chimiques qui ont lieu dans une pile d'alimentation électrique, une batterie ou un accumulateur.
Principe
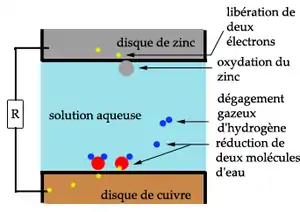
Pour avoir une corrosion galvanique, trois conditions sont nécessaires :
- des métaux de nature différente : c'est la différence de potentiel de dissolution entre les deux métaux qui provoque le phénomène. L'expérience montre qu'il faut une différence de potentiel de 100 mV pour voir apparaître la corrosion.
- la présence d'un électrolyte, en général aqueux : la présence d'ions dans le milieu aqueux (ex. : eau de mer), accélère le phénomène. Ce type de corrosion peut également exister dans un milieu anhydre mais ionique comme l'ammoniaque liquide.
- la continuité électrique entre les deux métaux : le phénomène diminue très rapidement en éloignant les deux métaux. Il faut qu'il y ait transfert de charges électriques pour avoir le phénomène de corrosion.
La masse de métal consommé est donnée par la loi de Faraday dans le cas d’un courant de corrosion constant :

- m : masse (g)
- M : masse molaire du métal
- z : valence (exemple : 3 pour l'aluminium)
- I : intensité électrique (A)
- t : temps (s)
- F : Constante de Faraday 96500 C environ
L'intensité est fonction de :
- la nature de l'électrolyte : elle augmente si le milieu est salin ;
- polarisation : certains produits de corrosion peuvent faire obstacle et ralentir ou bloquer les réactions chimiques ;
- la surface relative de la cathode et de l'anode. On peut établir le ratio suivant :

K augmente si la surface de la cathode augmente et/ou la surface de l'anode diminue. La corrosion galvanique sera donc très importante si l'on a une grande cathode et une petite anode.
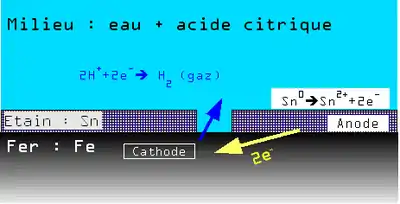
Exemple : cas des boîtes de conserve en tôle d'acier étamé
Une tôle étamée (fer-blanc) est une tôle d'acier sur laquelle a été appliquée une fine couche d'étain (Sn) pour la protéger.
Le revêtement peut présenter de légères discontinuités.
En présence d'eau le potentiel de dissolution du couple Fe2+/Fe est plus faible que celui du couple Sn2+/Sn. Il y a donc une très petite anode (Fe) et une très grande cathode (Sn) ce qui entraîne une corrosion galvanique rapide et une perforation de la boîte de conserve.
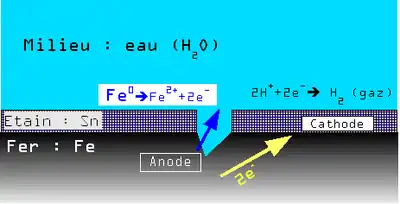
En présence d'acide citrique (fréquent dans les liquides alimentaires) par complexation de différents éléments chimiques, la position relative des couples Fe2+/Fe et Sn2+/Sn s'inverse.

Il y a donc dans ce cas une grande anode (Sn) et une petite cathode (Fe), et dissolution par corrosion de l'étain à l'intérieur de la boîte. Mais compte tenu du rapport de surface, cette dissolution est extrêmement lente et sur une très grande surface, ce phénomène assurant la durabilité de la boîte de conserve.
Corrosion par pile de concentration
La corrosion par pile de concentration est un cas très proche de la corrosion galvanique. La différence tient dans le fait qu'il y a corrosion sur une pièce de même métal. Il n'y a pas couplage de deux métaux de natures différentes. Seule la concentration du fluide qui baigne le métal varie.
Ce type de corrosion a lieu sur une même pièce. Elle a lieu lorsque la composition du milieu varie. En effet, le potentiel électrochimique est déterminé par le couple matériau/milieu, il suffit que l'un des deux varie pour que le potentiel varie. Par exemple, si une pièce dans un courant d'eau présente une cavité, l'eau dans cette cavité stagne et sa composition évolue avec les réactions chimiques d'oxydo-réduction ; par ailleurs, l'eau à l'extérieur de la cavité est continuellement renouvelée et garde la même composition, on peut donc avoir une pile qui se crée entre la cavité et l'extérieur de la pièce, donc une corrosion accélérée.
On voit ici qu'une même pièce se comportera de manière différente en eau stagnante et en eau mouvante. Si une canalisation présente un coude franc, le liquide à l'extérieur du coude est moins agité, il stagne, tandis que le liquide à l'intérieur du coude est agité, ce qui peut aussi produire une pile.
Généralités
La corrosion par aération différentielle se produit lorsqu'un même matériau est en contact avec deux milieux de teneurs en oxydant différentes. Par exemple, si un piquet est planté dans la terre ou la vase, la partie proche de la surface est en contact avec plus de dioxygène que la partie profonde, il peut donc se créer une pile entre la partie profonde et la partie en surface. La corrosion par aération différentielle se rencontre aussi pour les pièces immergées, lorsque la concentration en dioxygène évolue avec la profondeur. C'est parfois le cas des coques de bateaux. La couche d'eau proche de la surface est plus riche en oxygène que les couches profondes. Il peut y avoir corrosion au niveau de la ligne de flottaison.
Le problème d'aération différentielle peut se poser lorsqu'une pièce n'est peinte qu'en partie, ou lorsque la peinture est rayée.
L'effet Evans
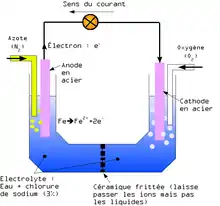
L'existence de la corrosion par aération différentielle a été démontrée en réalisant l'expérience suivante :
Deux électrodes en acier strictement identiques sont plongées dans un bain salin (avec du chlorure de sodium par exemple). Ce bain est séparé en deux moitiés par une céramique qui laisse passer les ions mais pas les molécules. Comme pour une pile, les deux électrodes sont reliées par un conducteur électrique. Dans une moitié du bain, on fait barboter de l'oxygène et dans l'autre de l'azote. Un courant électrique apparaît alors, la cathode étant du côté où l'eau est plus riche en oxygène.
Corrosion par différence de concentration
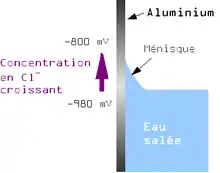
Le même phénomène est observable pour des différences de concentration d'autres éléments. On aura le même phénomène que pour la pile d'Evans en supprimant le barbotage de gaz et en ayant des concentrations différentes de cations métalliques dans chaque compartiment.
Pour les tôles d'aluminium immergées, c'est la différence de concentration en ion chlorure entre le ménisque (où l'évaporation est plus importante) et l'eau plus profonde qui peut provoquer la corrosion. Le phénomène étant renforcé par le fait que les potentiels de dissolution sont plus importants si la lame d'eau est plus faible[1]. L'apparition de corrosion dans ce cas dépend des alliages. L'expérience montre que certains alliages ne présentent pas de corrosion dans ce cas. Cette corrosion est d'ailleurs parfois appelée corrosion à la ligne de surface.
Les piqûres
Généralités
La formation de piqûres (pitting) est un phénomène de corrosion qui survient lorsqu'une pièce est protégée contre la corrosion généralisée par un film passif, souvent un oxyde protecteur (par exemple, acier inoxydable ou aluminium).
Il s'agit d'une corrosion localisée. En surface, on ne voit qu'un petit point, mais en dessous, il y a une cavité bien plus importante. Ceci entraîne à terme la perforation de la pièce et sa fragilisation.
La formation de piqûres est un phénomène d'autant plus redouté qu'il surprend l'utilisateur : celui-ci a pensé à la corrosion généralisée, il pense être protégé, et la trace extérieure de corrosion est quasiment indétectable.
Mécanisme général
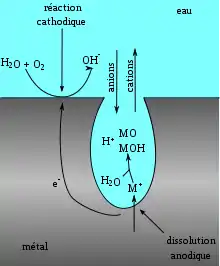
La piqûre commence toujours par une rupture locale du film passif (l'oxyde protecteur formé sur le métal) souvent au droit d'hétérogénéités du métal près desquelles le film est moins stable — ex. : inclusions de type sulfure (MnS, NiS) pour les aciers inoxydables ou que le métal soit en présence de chlorures ou de thiosulfates (issus de l'oxydation de sulfures) qui sont des ions perforants. Ensuite, la propagation est entretenue par deux phénomènes :
- l'intérieur de la piqûre est dépassivé et contient un milieu désaéré, alors que l'extérieur est passivé et en milieu aéré. Il se crée donc un effet de pile entre l'intérieur et l'extérieur, il s'agit en fait d'une corrosion galvanique localisée ;
- La réduction de l'oxygène à l'extérieur de la piqûre, sur le métal passif, alimente l'oxydation du métal à l'intérieur, ce qui produit des cations métalliques. Lorsque la concentration en cations augmente se produit une réaction d'hydrolyse, par exemple pour les aciers inoxydables : Cr3+ + 3 H2O → Cr(OH)3 + 3 H+.
On voit que cette réaction d'hydrolyse libère de l'acidité. La production de cations H+ va entraîner la migration d'anions à l'intérieur de la piqûre afin de rétablir la neutralité électrique, généralement des chlorures (ou des thiosulfates dans le cas de l'oxydation des sulfures), agressifs pour le métal. L'addition de ces deux phénomènes entraîne donc la formation d'un milieu acide et concentré en chlorures, qui à son tour accélère la propagation de la piqûre. Pour les aciers inoxydables, des milieux de pH 0 et de concentration en chlorures d'une mole par litre ont pu être ainsi identifiés dans les piqûres, alors que le milieu externe était proche de la neutralité (pH 7) et peu chargé en chlorures.
Lorsque la corrosion est également liée à l'oxydation de sulfures (oxydation de la pyrite en milieu argileux, par exemple), il se forme de façon transitoire des ions thiosulfates (S2O32−) particulièrement agressifs, car étant toujours doublement chargés négativement (base conjuguée d'un acide fort) et ainsi dotés d'une conductivité électrique équivalente supérieure à celles des ions chlorures. Cela signifie donc, que les anions thiosulfates migrent dans les piqûres encore plus rapidement que les ions chlorures afin d'y rétablir l'électroneutralité (pour compenser la formation de cations Men+, comme Fe2+). En effet, en électrophorèse capillaire, la vitesse d'électromigration évolue dans l'ordre suivant d'électromobilité: H+ > OH− > S2O32− > Cl−.
Cependant, au bout d'un certain temps, la corrosion par piqûre ne s'accélère plus car la cinétique devient contrôlée par la diffusion-migration des anions provenant de l'extérieur, alors que les chemins à parcourir s'allongent en raison de la croissance de la profondeur de la piqûre.
Le phénomène
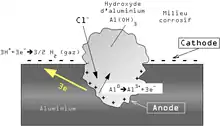

L'aluminium est naturellement recouvert d'une couche de protection, ou couche de passivation. Il s'agit d'une couche d'oxyde formée suivant la réaction :

La très forte réactivité de l'aluminium est due à une valeur élevée de l'énergie libre (-1675 kJ)
La piqûration est provoquée par la rupture du film d'oxyde dans un milieu contenant, par exemple, des ions chlorure (Cl−). La piqûration est influencée par le milieu dans lequel se trouve l'aluminium : acide, nourriture. La composition de l'alliage peut également entrer en ligne de compte. La présence de cuivre dans l'alliage peut par exemple être une cause de piqûration. Cependant, le milieu extérieur est le facteur dominant[3]
Après la rupture du film protecteur, l'aluminium nu devient une anode où se produit la réaction d'oxydation suivante :

À la cathode sur la surface du métal, nous avons les réactions suivantes :


Globalement, l'aluminium métal se dissout pour former de l'hydroxyde d'aluminium (souvent appelé par erreur alumine dans le langage courant) suivant la réaction :

| Aspect de surface d'un alliage d'aluminium type 7000 avec des piqûres de corrosion. | Aspect de surface d'un alliage d'aluminium type 7000 avec des piqûres de corrosion (après décapage) | Piqûre (gros plan) |
|---|---|---|
 Piqûres avec l'hydroxyde d'aluminium. |
 Piqûre sans l'hydroxyde. |
 Gros plan, la pustule d'hydroxyde d'aluminium est bien visible. |
Vitesse de propagation de la piqûre
La profondeur de la piqûre augmente rapidement au départ, puis la vitesse d'augmentation ralentit avec le temps[1] :
![d=k{\sqrt[ {3}]{t}}\,\!](https://img.franco.wiki/i/076efc4d962aedea24f7773c3d0a479c4980d122.svg)
- d: profondeur de la piqûre
- t : temps
- k : constante dépendant de l'alliage et des conditions (température, nature du fluide etc.)
En effet, dans le cas d'une piqûre hémisphérique de rayon r idéale, la quantité de métal dissous pendant un temps t est :

Corrosion microbienne
Outre les propriétés chimiques et physiques de l'environnement, l'activité microbienne peut également jouer un rôle important dans les processus de corrosion.
Le métabolisme des micro-organismes peut modifier localement la composition chimique à l'interface de la pièce, et donc créer une corrosion localisée. C'est notamment le cas des bactéries sulfato-réductrices (BSR). De grandes concrétions de rouille, comme celles observées sur l'épave du Titanic, les rusticles, peuvent être formées par des communautés de bactéries halophiles anaérobies dont le métabolisme énergétique est basé sur l'oxydation du fer métallique.
Autres interactions avec les organismes vivants
Les mollusques, qui se fixent sur les coques de bateaux et les piliers immergés, peuvent poser problème. Les peintures anti-fouling à base de composés organo-étains longtemps utilisées et très efficaces ont dû être abandonnées en raison de leur trop grande toxicité environnementale.
La protection contre la corrosion peut aussi avoir un impact négatif sur l'environnement, en libérant des substances toxiques. C'est ainsi que les peintures anti-rouille au minium à base d'oxydes mixtes de plomb (Pb3O4, ou PbO2 · 2 PbO) ont été abandonnées. La teneur en zinc due à la dissolution des anodes sacrificielles peut aussi poser problème, c'est la raison pour laquelle des solutions de protection cathodiques par courant imposé (courant contraire) sont préférables.
Le problème de l'interaction entre le métal et le milieu vivant se pose aussi dans le cas d'implants et de prothèses, notamment pour les soins dentaires (plombages, couronnes), les prothèses articulaires (prothèse totale de hanche), et les broches, plaques et vis posées à titre provisoire ou permanent pour soigner certaines fractures et luxations. Pour les prothèses articulaires sollicitées par des contraintes mécaniques importantes on utilise en général comme matériau des aciers inoxydables ou du titane.
Corrosion par courants vagabonds
C'est une autre forme de corrosion qui se traduit par la déperdition au fil du temps des structures métalliques autour des chemins de fer à motion électrique.
Phénomènes d'interface
La corrosion est un phénomène d'interface (contact entre la surface de la pièce et l'environnement). Tout ce qui modifie les propriétés de l'interface peut avoir une influence sur la corrosion.
Notamment, la circulation d'eau provoque un renouvellement de la solution, et modifie le transfert des espèces dans la solution (diffusion). La présence de particules (par exemple grains de sable) peut créer une érosion qui raye la couche protectrice (corrosion-érosion). Si l'on met la pièce en extension, on modifie l'énergie d'interface, donc l'adsorption des espèces, donc les réactions chimiques pouvant avoir lieu (corrosion sous contrainte).
Les méthodes d'étude de la corrosion aqueuse
Études en laboratoire
Nous avons vu que les principaux facteurs de la corrosion des métaux en milieu aqueux sont la température, le pH et le potentiel électrochimique, et que la corrosion implique une circulation d'électrons. L'étude de la corrosion va donc se faire par suivi du pH avec un pH-mètre et de la température avec un thermomètre, ou bien en les imposant en maîtrisant la composition de la solution et sa température pour des études ex situ (c'est-à-dire en laboratoire).
Pour le suivi du potentiel, on utilise un voltmètre relié d'un côté à la pièce métallique, et de l'autre à une électrode de référence, en général une électrode au calomel saturée (« ECS ») — en fait, en raison de la précision nécessaire, on utilise un appareil appelé « potentiomètre » qui possède d'autres fonctions. Ce montage permet de mesurer le « potentiel d'abandon », c'est-à-dire le potentiel pris naturellement par la pièce étudiée par rapport à la solution, on peut ainsi déterminer si la pièce est passivée ou au contraire se corrode. Le suivi du courant permet de mesurer la vitesse de corrosion. Pour cela, il faut une troisième électrode. On utilise ainsi une électrode inerte, par exemple en graphite ou en platine.
Outre ces méthodes passives, on peut imposer un potentiel ou un courant et voir comment le système se comporte. En effet, les phénomènes de corrosion mettent en œuvre des réactions d'oxydo-réduction, mais aussi des phénomènes de diffusion et d'adsorption. On peut modéliser la relation entre le courant de corrosion et le potentiel par un circuit RLC ; ainsi, en envoyant des perturbations sinusoïdales et en regardant la réponse du système, on peut définir une impédance Z, et déduire de cette impédance les phénomènes qui ont lieu.
Pour simuler la circulation d'eau, on peut bien sûr employer une pompe pour créer un flux laminaire, mais on peut aussi faire tourner la pièce, la vitesse de rotation symbolisant la vitesse de circulation de l'eau. On s'éloigne ainsi des conditions « réelles », mais en revanche, on maîtrise mieux la vitesse, et l'on peut ainsi envoyer des perturbations sinusoïdales à la vitesse de rotation et déterminer une impédance de rotation (c'est-à-dire regarder la variation de potentiel et de courant en fonction de la variation de la vitesse), ce qui donne accès à d'autres paramètres, notamment à ce qui concerne la diffusion dans le liquide.
Les techniques perturbatrices ont un inconvénient majeur : il faut imposer les conditions de potentiel au système, de ce fait, il ne se comporte pas de manière « naturelle ». Pour éviter cela, on peut mesurer les fluctuations naturelles du potentiel et du courant, ce que l'on appelle le « bruit électrochimique », et en déduire de même une impédance.
Les méthodes ci-dessus sont globales, elles donnent des valeurs moyennes sur la pièce. Or, on sait que les phénomènes de corrosion sont très souvent localisés. Pour mesurer le potentiel, on peut utiliser une électrode ayant la forme d'une fine aiguille et faire une cartographie de potentiel. On peut ainsi détecter des zones anodiques et des zones cathodiques sur une pièce, par exemple dans les phénomènes de piqûration. Cette technique est appelée SRET (scanning reference electrode technique), ou SVET (scanning vibrating electrode technique) si la pointe vibre.
Certains phénomènes produisent des sons, par exemple la création de bulles d'hydrogène. On peut aussi faire un suivi de corrosion en enregistrant ces sons.
Suivi in situ
Il est aussi nécessaire de suivre la corrosion sur des installations industrielles ou portuaires, afin d'effectuer de la maintenance préventive et d'éviter les accidents. Outre l'inspection visuelle, on peut suivre in situ l'évolution du potentiel et ou du courant et en déduire l'état de la pièce. Mais on peut aussi utiliser des mesures macroscopiques, comme des mesures d'épaisseur de pièces (pièces fonctionnelles ou témoins de corrosion). On peut aussi chercher des cavités (par exemple des piqûres) avec des ultrasons. Pour les pipelines et les forages profonds, on utilise des outils de contrôle internes automatiques comme les racleurs intelligents [4] et les sondes de diagraphies[5].
Protection contre la corrosion aqueuse
Pour protéger les métaux corrodables mouillés, on a le choix d'isoler ces métaux de l'eau avec de la peinture, de les recouvrir de matière plastique ou d'effectuer une oxydation anodique chromique colmatée (alliage d'aluminium uniquement). On peut aussi utiliser la protection cathodique soit avec une anode sacrificielle dite « anode réactive », soit en utilisant un dispositif à courant imposé.
Notes et références
- Corrosion de l'aluminium, Christian Vargel, 2d. Dunod, 2002, (ISBN 2-10-006569-6).
- Le comportement de l'aluminium et de ses alliages, Christian Vargel, Dunod 1979, (ISBN 2-04-010078-4)
- Aluminium Viewed from Within, D.Altenpohl, Aluminium-Verlag, Düsseldorf, 1982, (ISBN 3-87017-138-3)
- Stéphane Sainson, Inspection en ligne des pipelines. Principes et méthodes, Éd. Lavoisier, 2007 (ISBN 978-2743009724), 332 p.
- Stéphane Sainson, Les diagraphies de corrosion. Acquisition et interprétation des données, Éd. Lavoisier, 2010 (ISBN 978-2743012014), 547 p.
Annexes
Articles connexes
Bibliographie
- Corrosion 5e éd., vol. 13 de ASM Handbook, éd. ASM International (American Society for Materials), 1996.
- Corrosion et chimie de surfaces des métaux, D. Landolt, vol. 12 de Traité des matériaux, Presses polytechniques et universitaires romandes, 1993.
- Métallurgie, du minerai au matériau, J. Philibert et coll., éd. Masson, 1998.
- Corrosion de l'aluminium, Christian Vargel, 2d. Dunod, 2002, (ISBN 2-10-006569-6).
- Le comportement de l'aluminium et de ses alliages, Christian Vargel, Dunod, 1979, (ISBN 2-04-010078-4).
- Aluminium Viewed from Within, D.Altenpohl, Aluminium-Verlag, Düsseldorf, 1982, (ISBN 3-87017-138-3).