Protéinopathie
En médecine, les protéinopathies font référence à une classe de maladies dans lesquelles certaines protéines deviennent structurellement anormales et perturbent ainsi la fonction des cellules, des tissus et des organes du corps[1] - [2]. Souvent, les protéines ne parviennent pas à se replier dans leur configuration normale ; dans cet état mal replié, les protéines peuvent devenir toxiques (en acquérant de nouvelles fonctions indésirées par mutation) ou perdre leur fonction normale[3]. Les protéinopathies (également connues sous le nom de troubles de conformation des protéines ou des maladies de repliement protéique) comprennent des maladies telles que la maladie de Creutzfeldt-Jakob et autres maladies à prions, la maladie d'Alzheimer, de Parkinson , les amyloses, l'atrophie multisystématisée, et un large éventail d'autres troubles[4] - [5] - [6] - [7] - [8]. Le terme anglais proteopathy a été proposé pour la première fois en 2000 par Lary Walker et Harry LeVine[1].
Le concept de protéinopathie trouver ses origines au milieu du XIXe siècle, lorsque, en 1854, Rudolf Virchow a inventé le terme amyloïde («amylacé») pour décrire une substance dans les corps cérébraux amylacées qui présentait une réaction chimique ressemblant à celle de la cellulose . En 1859, Friedreich et Kekulé ont démontré que, plutôt que de se composer de cellulose, « l'amyloïde » est en fait riche en protéines[9]. Des recherches ultérieures ont montré que de nombreuses protéines différentes peuvent former des substances amyloïdes et que toutes les substances amyloïdes présentent une biréfringence en lumière polarisée croisée après coloration avec le colorant rouge Congo, ainsi qu'une ultrastructure fibrillaire vue au microscope électronique . Cependant, certaines lésions protéiniques sont dépourvues de biréfringence et contiennent peu ou pas de fibrilles amyloïdes classiques, comme les dépôts diffus de protéine bêta (Aβ) amyloïde dans le cerveau des personnes atteintes de la maladie d'Alzheimer[10]. En outre, il a été mis en évidence que de petits agrégats de protéines non fibrillaires connus sous le nom d'oligomères seraient toxiques pour les cellules d'un organe affecté, et que les protéines amyloïdogènes sous leur forme fibrillaire pouvaient être relativement bénignes[11] - [12].
Mécanisme biochimique
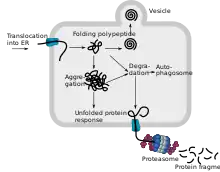
Dans la plupart des cellules de tous les organismes, les protéines les plus diverses sont constamment produites au cours de la synthèse des protéines, qui remplissent une grande variété de fonctions dans la cellule et dans tout l'organisme. Pour qu'une protéine fonctionne correctement, sa structure tertiaire est d'une importance cruciale. Cette structure est obtenue grâce à un processus appelé repliement des protéines. Le repliement des protéines est un processus complexe et délicat. Le contrôle de la qualité des protéines est responsable du repliement correct des protéines supervisé. Statistiquement, environ 30 % de toutes les protéines issues de la biosynthèse des protéines sont incorrectement repliées et sont normalement décomposées dans le protéasome de la cellule en une dizaine de minutes[13] - [14]. L'accumulation de protéines incorrectement repliées dans le réticulum endoplasmique conduit, en réponse au stress engendré dans le RE, à un signal de réponse des protéines dépliées (UPR), associées notamment à une suppression de la traduction et à une synthèse accrue de chaperons[15] .
Une seule molécule de protéine mal repliée n'est pas responsable des images cliniques graves de maladies de mauvais repliement des protéines. Pour que cela se produise, de grandes quantités de ces protéines doivent être produites ou le nombre de molécules correctement repliées doit être réduit. Dans le cas des prions, cela se produit parce qu'une molécule mal repliée au contact d'une molécule correctement pliée provoque le dépliement de la molécule « correcte ».
Les protéines défectueuses sont également appelées produits ribosomaux défectueux ((en) Defective ribosomal products ou DRiPs)[16].
Physiopathologie
Dans la plupart des protéinopathies, sinon toutes, un changement du repliement tridimensionnel (conformation) augmente la tendance d'une protéine spécifique à se lier à elle-même[5]. Sous cette forme agrégée, la protéine résiste à la clairance et peut interférer avec la capacité normale des organes affectés. Dans certains cas, un mauvais repliement de la protéine entraîne une perte de sa fonction habituelle. Par exemple, la mucoviscidose est causée par une anomalie de la protéine CFTR[3] et dans la sclérose latérale amyotrophique et la dégénérescence lobaire frontotemporale (FTLD), certaines protéines de régulation génique s'agrègent de manière inappropriée dans le cytoplasme, et sont donc incapables d'accomplir leurs tâches normales au sein du noyau[17] - [18]. Les protéines partageant une caractéristique structurelle commune connue sous le nom de squelette polypeptidique, elles ont toutes le potentiel de mal se replier dans certaines circonstances[19]. Cependant, seul un nombre relativement faible de protéines est lié à des troubles protéopathiques, probablement en raison de particularités structurelles des protéines vulnérables. Par exemple, les protéines qui sont normalement dépliées ou relativement instables en tant que monomères sont plus susceptibles de se replier en une conformation anormale[20]. Dans presque tous les cas, la configuration moléculaire causant la maladie implique une augmentation des feuillets bêta dans la structure secondaire de la protéine[21] - [22]. Il a été démontré que les protéines anormales dans certaines protéinopathies peuvent se replier sous différentes formes tridimensionnelles ; ces structures protéiniques variantes sont définies par leurs différentes propriétés pathogènes, biochimiques et conformationnelles[23]. Elles ont été particulièrement étudiées dans le cadre des maladies à prions et sont appelées souches protéiques[24] - [25]
_of_alpha-synuclein_in_Lewy_Bodies_and_Lewy_Neurites_in_the_neocortex_of_a_patient_with_Lewy_Body_Disease.jpg.webp)
La probabilité qu'une protéinopathie se développe est augmentée par certains facteurs de risque qui favorisent l'auto-assemblage d'une protéine. Ceux-ci incluent des changements déstabilisants dans la séquence d'acides aminés primaire de la protéine, des modifications post-traductionnelles (telles que l'hyperphosphorylation ), des changements de température ou de pH, une augmentation de la production d'une protéine ou une diminution de sa clairance [1] - [26] - [27]. L'âge avancé est un facteur de risque important comme le traumatisme crânien[28] - [29]. Dans le cerveau vieillissant, plusieurs protéinopathies peuvent se cumuler[30] Par exemple, en plus de la tauopathie et de l'Aβ-amyloïdose (qui coexistent en tant que caractéristiques pathologiques clés de la maladie d'Alzheimer), de nombreux patients atteints d'Alzheimer présentent une synucléinopathie concomitante (corps de Lewy) dans le cerveau[31]
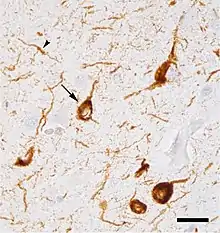
On suppose que les chaperons et les co-chaperons (protéines qui aident au repliement des protéines) peuvent agir comme antagonistes de la protéinotoxicité pendant le vieillissement et dans les maladies de mauvais repliement des protéines pour maintenir la protéinostase[32] - [33] - [34].
Contamination
Certaines protéines peuvent être amenées à former des assemblages anormaux par exposition à une protéine identique (ou similaire) qui s'est repliée dans une conformation causant la maladie, un processus appelé « ensemencement » ou « modèle permissif »[35] - [36]. De cette manière, l'état pathologique peut être provoqué chez un hôte sensible par l'introduction d'un extrait de tissu malade d'un donneur porteur. La forme la plus connue d'une telle protéinopathie inductible est la maladie à prion[37], qui peut être transmise par exposition d'un organisme hôte à une protéine prion purifiée dans une conformation pathogène[38] - [39]. Il existe maintenant des preuves que d'autres protéinopathies peuvent être induites par un mécanisme similaire, y compris l'amylose Aβ , l'amylose A (AA) , l'amylose apolipoprotéine AII[40], les tauopathies[41] - [42] la synucléinopathie[43] - [44] - [45] - [46], et les agrégations de superoxyde dismutase -1 (SOD1)[47] - [48], de polyglutamine[49] - [50], et de TAR DNA-binding protein-43 (TDP-43)[51]. Pour toutes ces raisons, Stanley Prusiner suggère de ne pas utiliser le terme de graine protinéopathique, mais d'englober toutes les protéines agrégées qui adoptent des formes alternatives et subissent une auto-propagation sous la nomenclature globale de prions[52].
Dans tous ces cas, une forme aberrante de la protéine elle-même semble être l'agent pathogène. Dans certains cas, le dépôt d'un type de protéine peut être induit expérimentalement par des assemblages agrégés d'autres protéines riches en structure en feuillet β, probablement en raison de la complémentarité structurelle des molécules protéiques. Par exemple, l'amylose AA peut être stimulée chez la souris par des macromolécules aussi diverses que la soie, l'amyloïde de levure Sup35 et les fibrilles de curli de la bactérie Escherichia coli [53]. L'amyloïde AII peut être induite chez la souris par une variété de fibrilles amyloïdes riches en feuillets β[54], et les tauopathies cérébrales peuvent être induites par des extraits de cerveau riches en Aβ agrégé[55]. Il existe également des preuves expérimentales d'un ensemencement croisé entre la protéine prion et l'Aβ[56]. En général, un tel ensemencement hétérologue est moins efficace que l'ensemencement par une forme corrompue de la même protéine.
Liste de protéinopathies
Groupe des amyloses
Il existe différentes protéines concernées dans le groupe des amyloses— qui sont désormais préférentiellement désignées sous la forme « A + nom de la protéine précurseur »[72]. L'amylose AL (en) est associée à des fragments d'immunoglobulines appelés chaîne légère (en). L'amylose familiale de type finnois (en), une maladie rare identifiée chez 400 à 600 Finlandais, résulte de la protéolyse aberrante d'une forme mutante de la gelsoline plasmatique (en) (pGSN)[84].
Traitements
Le développement de traitements efficaces pour de nombreuses protéinopathies est un défi[88] - [89]. Parce que les protéopathies impliquent souvent différentes protéines provenant de différentes sources, les stratégies de traitement doivent être adaptées à chaque trouble ; cependant, les approches thérapeutiques générales comprennent le maintien de la fonction des organes affectés, la réduction de la formation des protéines pathogènes, la prévention du mauvais repliement ou de l'agrégation des protéines, ou la favorisation de leur élimination[90] - [91]. Par exemple, dans la maladie d'Alzheimer, les chercheurs cherchent des moyens de réduire la production de la protéine Aβ associée à la maladie en inhibant les enzymes qui la libèrent de sa protéine mère. Une autre stratégie consiste à utiliser des anticorps pour neutraliser des protéines spécifiques par immunisation active ou passive[92]. Dans certaines protéinopathies, l'inhibition des effets toxiques des oligomères protéiques peut être bénéfique[93]. L'amylose A (AA) peut être réduite en traitant l'état inflammatoire qui augmente la quantité de protéines dans le sang (appelée amyloïde sérique A ou SAA). Dans l'amylose à chaînes légères d'immunoglobulines (amylose AL), la chimiothérapie peut être utilisée pour réduire le nombre de cellules sanguines qui fabriquent la protéine de chaîne légère qui forme l'amyloïde dans divers organes[94]. L'amylose à transthyrétine (ATTR) résulte du dépôt de transthyrétine (TTR) mal repliée dans plusieurs organes[95]. Étant donné que la TTR est principalement produite dans le foie, l'amylose à TTR peut être ralentie dans certains cas héréditaires par transplantation hépatique[96]. L'amylose à TTR peut également être traitée en stabilisant les assemblages normaux de la protéine (appelés tétramères car ils se composent de quatre molécules TTR liées ensemble). La stabilisation empêche les molécules de TTR individuelles de s'échapper, de mal se replier et de s'agréger en fibres amyloïde[97] - [98]
Plusieurs autres stratégies de traitement des protéinopathies sont à l'étude, notamment des petites molécules et des médicaments biologiques tels que de petits ARN interférents, des oligonucléotides antisens, des peptides et des cellules immunitaires modifiées[97] - [94] - [99] - [100]. Dans certains cas, plusieurs agents thérapeutiques peuvent être combinés pour améliorer l'efficacité[101].
Références
- (en) LC Walker et H. LeVine, « The cerebral proteopathies », Neurobiology of Aging, vol. 21, no 4, , p. 559–61 (PMID 10924770, DOI 10.1016/S0197-4580(00)00160-3)
- (en) LC Walker et H. LeVine, « The cerebral proteopathies: neurodegenerative disorders of protein conformation and assembly », Molecular Neurobiology, vol. 21, nos 1–2, , p. 83–95 (PMID 11327151, DOI 10.1385/MN:21:1-2:083)
- (en) LM Luheshi, DC Crowther et CM Dobson, « Protein misfolding and disease: from the test tube to the organism », Current Opinion in Chemical Biology, vol. 12, no 1, , p. 25–31 (PMID 18295611, DOI 10.1016/j.cbpa.2008.02.011)
- (en) Fabrizio Chiti et Christopher M. Dobson, « Protein Misfolding, Functional Amyloid, and Human Disease », Annual Review of Biochemistry, vol. 75, no 1, , p. 333–366 (ISSN 0066-4154 et 1545-4509, DOI 10.1146/annurev.biochem.75.101304.123901, lire en ligne, consulté le )
- (en) Robin W Carrell et David A Lomas, « Conformational disease », The Lancet, vol. 350, no 9071, , p. 134–138 (DOI 10.1016/S0140-6736(97)02073-4, lire en ligne, consulté le )
- (en) Per Westermark, Merrill D. Benson, Joel N. Buxbaum et Alan S. Cohen, « A primer of amyloid nomenclature », Amyloid, vol. 14, no 3, , p. 179–183 (ISSN 1350-6129 et 1744-2818, DOI 10.1080/13506120701460923, lire en ligne, consulté le )
- (en) Gunilla T. Westermark, Marcus Fändrich, Katarzyna Lundmark et Per Westermark, « Noncerebral Amyloidoses: Aspects on Seeding, Cross-Seeding, and Transmission », Cold Spring Harbor Perspectives in Medicine, vol. 8, no 1, , a024323 (ISSN 2157-1422, PMID 28108533, PMCID PMC5749146, DOI 10.1101/cshperspect.a024323, lire en ligne, consulté le )
- (en) Stanley B. Prusiner, « Biology and Genetics of Prions Causing Neurodegeneration », Annual Review of Genetics, vol. 47, no 1, , p. 601–623 (ISSN 0066-4197 et 1545-2948, PMID 24274755, PMCID PMC4010318, DOI 10.1146/annurev-genet-110711-155524, lire en ligne, consulté le )
- (en) Jean D. Sipe et Alan S. Cohen, « Review: History of the Amyloid Fibril », Journal of Structural Biology, vol. 130, nos 2-3, , p. 88–98 (DOI 10.1006/jsbi.2000.4221, lire en ligne, consulté le )
- (en) Henryk M. Wisniewski, Marcin Sadowski, Katarzyna Jakubowska-Sadowswka et Michal Tarnawski, « Diffuse, Lake-like Amyloid-β Deposits in the Parvopyramidal Layer of the Presubiculum in Alzheimer Disease: », Journal of Neuropathology and Experimental Neurology, vol. 57, no 7, , p. 674–683 (ISSN 0022-3069, DOI 10.1097/00005072-199807000-00004, lire en ligne, consulté le )
- (en) Charles G. Glabe, « Common mechanisms of amyloid oligomer pathogenesis in degenerative disease », Neurobiology of Aging, vol. 27, no 4, , p. 570–575 (DOI 10.1016/j.neurobiolaging.2005.04.017, lire en ligne, consulté le )
- Bharathi Shrikanth Gadad, Gabrielle B. Britton et K.S. Rao, « Targeting Oligomers in Neurodegenerative Disorders: Lessons from α-Synuclein, Tau, and Amyloid-β Peptide », Journal of Alzheimer's Disease, vol. 24, no s2, , p. 223–232 (DOI 10.3233/JAD-2011-110182, lire en ligne, consulté le )
- (en) Douglas M. Cyr et Daniel N. Hebert, « Protein quality control—linking the unfolded protein response to disease: Conference on ‘From Unfolded Proteins in the Endoplasmic Reticulum to Disease’ », EMBO reports, vol. 10, no 11, , p. 1206–1210 (ISSN 1469-221X et 1469-3178, PMID 19851332, PMCID PMC2775177, DOI 10.1038/embor.2009.224, lire en ligne, consulté le )
- (en) Shu-Bing Qian, Jack R. Bennink et Jonathan W. Yewdell, « Quantitating Defective Ribosome Products », dans Ubiquitin-Proteasome Protocols, vol. 301, Humana Press, (ISBN 978-1-59259-895-3, DOI 10.1385/1-59259-895-1:271, lire en ligne), p. 271–282
- Saïd Taouji et Éric Chevet, « Modulation pharmacologique de la réponse au stress du réticulum endoplasmique: Potentiel thérapeutique en cancérologie », médecine/sciences, vol. 31, nos 6-7, , p. 667–673 (ISSN 0767-0974 et 1958-5381, DOI 10.1051/medsci/20153106021, lire en ligne, consulté le )
- J. W. Yewdell, L. C. Antón et J. R. Bennink, « Defective ribosomal products (DRiPs): a major source of antigenic peptides for MHC class I molecules? », Journal of Immunology (Baltimore, Md.: 1950), vol. 157, no 5, , p. 1823–1826 (ISSN 0022-1767, PMID 8757297, lire en ligne, consulté le )
- (en) D. Ito et N. Suzuki, « Conjoint pathologic cascades mediated by ALS/FTLD-U linked RNA-binding proteins TDP-43 and FUS », Neurology, vol. 77, no 17, , p. 1636–1643 (ISSN 0028-3878 et 1526-632X, PMID 21956718, PMCID PMC3198978, DOI 10.1212/WNL.0b013e3182343365, lire en ligne, consulté le )
- Benjamin Wolozin et Daniel Apicco, « RNA Binding Proteins and the Genesis of Neurodegenerative Diseases », dans GeNeDis 2014, vol. 822, Springer International Publishing, (ISBN 978-3-319-08926-3, PMID 25416971, PMCID PMC4694570, DOI 10.1007/978-3-319-08927-0_3, lire en ligne), p. 11–15
- (en) Christopher M. Dobson, « Protein misfolding, evolution and disease », Trends in Biochemical Sciences, vol. 24, no 9, , p. 329–332 (DOI 10.1016/S0968-0004(99)01445-0, lire en ligne, consulté le )
- (en) Mathias Jucker et Lary C. Walker, « Self-propagation of pathogenic protein aggregates in neurodegenerative diseases », Nature, vol. 501, no 7465, , p. 45–51 (ISSN 0028-0836 et 1476-4687, PMID 24005412, PMCID PMC3963807, DOI 10.1038/nature12481, lire en ligne, consulté le )
- (en) Dennis J. Selkoe, « Folding proteins in fatal ways », Nature, vol. 426, no 6968, , p. 900–904 (ISSN 0028-0836 et 1476-4687, DOI 10.1038/nature02264, lire en ligne, consulté le )
- (en) David Eisenberg et Mathias Jucker, « The Amyloid State of Proteins in Human Diseases », Cell, vol. 148, no 6, , p. 1188–1203 (PMID 22424229, PMCID PMC3353745, DOI 10.1016/j.cell.2012.02.022, lire en ligne, consulté le )
- (en) Lary C. Walker, « Proteopathic Strains and the Heterogeneity of Neurodegenerative Diseases », Annual Review of Genetics, vol. 50, no 1, , p. 329–346 (ISSN 0066-4197 et 1545-2948, PMID 27893962, PMCID PMC6690197, DOI 10.1146/annurev-genet-120215-034943, lire en ligne, consulté le )
- (en) J. Collinge et A. R. Clarke, « A General Model of Prion Strains and Their Pathogenicity », Science, vol. 318, no 5852, , p. 930–936 (ISSN 0036-8075 et 1095-9203, DOI 10.1126/science.1138718, lire en ligne, consulté le )
- (en) David W. Colby et Stanley B. Prusiner, « De novo generation of prion strains », Nature Reviews Microbiology, vol. 9, no 11, , p. 771–777 (ISSN 1740-1526 et 1740-1534, PMID 21947062, PMCID PMC3924856, DOI 10.1038/nrmicro2650, lire en ligne, consulté le )
- (en) Robin W Carrell et David A Lomas, « Conformational disease », The Lancet, vol. 350, no 9071, , p. 134–138 (DOI 10.1016/S0140-6736(97)02073-4, lire en ligne, consulté le )
- (en) CM Dobson, « Protein misfolding, evolution and disease », Trends in Biochemical Sciences, vol. 24, no 9, , p. 329–32 (PMID 10470028, DOI 10.1016/S0968-0004(99)01445-0)
- (en) Steven T. DeKosky, Milos D. Ikonomovic et Sam Gandy, « Traumatic Brain Injury — Football, Warfare, and Long-Term Effects », New England Journal of Medicine, vol. 363, no 14, , p. 1293–1296 (ISSN 0028-4793 et 1533-4406, DOI 10.1056/NEJMp1007051, lire en ligne, consulté le )
- (en) McKee AC, Stein TD, Kiernan PT et Alvarez VE, « The neuropathology of chronic traumatic encephalopathy », Brain Pathology, vol. 25, no 3, , p. 350–64 (PMID 25904048, PMCID 4526170, DOI 10.1111/bpa.12248)
- (en) Peter T. Nelson, Irina Alafuzoff, Eileen H. Bigio, Constantin Bouras et al., « Correlation of Alzheimer Disease Neuropathologic Changes With Cognitive Status: A Review of the Literature », Journal of Neuropathology & Experimental Neurology, vol. 71, no 5, , p. 362–381 (ISSN 0022-3069 et 1554-6578, PMID 22487856, PMCID PMC3560290, DOI 10.1097/NEN.0b013e31825018f7, lire en ligne, consulté le )
- (en) Mrak RE et Griffin WS, « Dementia with Lewy bodies: Definition, diagnosis, and pathogenic relationship to Alzheimer's disease », Neuropsychiatric Disease and Treatment, vol. 3, no 5, , p. 619–25 (PMID 19300591, PMCID 2656298)
- (en) Peter M. Douglas, Daniel W. Summers et Douglas M. Cyr, « Molecular chaperones antagonize proteotoxicity by differentially modulating protein aggregation pathways », Prion, vol. 3, no 2, , p. 51–58 (ISSN 1933-6896 et 1933-690X, PMID 19421006, PMCID PMC2712599, DOI 10.4161/pri.3.2.8587, lire en ligne, consulté le )
- (en) Marc Brehme, Cindy Voisine, Thomas Rolland et Shinichiro Wachi, « A Chaperome Subnetwork Safeguards Proteostasis in Aging and Neurodegenerative Disease », Cell Reports, vol. 9, no 3, , p. 1135–1150 (DOI 10.1016/j.celrep.2014.09.042, lire en ligne, consulté le )
- (en) Marc Brehme et Cindy Voisine, « Model systems of protein-misfolding diseases reveal chaperone modifiers of proteotoxicity », Disease Models & Mechanisms, vol. 9, no 8, , p. 823–838 (ISSN 1754-8403 et 1754-8411, PMID 27491084, PMCID PMC5007983, DOI 10.1242/dmm.024703, lire en ligne, consulté le )
- (en) J. Hardy, « Expression of normal sequence pathogenic proteins for neurodegenerative disease contributes to disease risk: ‘permissive templating’ as a general mechanism underlying neurodegeneration », Biochemical Society Transactions, vol. 33, no 4, , p. 578–581 (ISSN 0300-5127 et 1470-8752, DOI 10.1042/BST0330578, lire en ligne, consulté le )
- (en) Lary C. Walker, Harry LeVine, Mark P. Mattson et Mathias Jucker, « Inducible proteopathies », Trends in Neurosciences, vol. 29, no 8, , p. 438–443 (DOI 10.1016/j.tins.2006.06.010, lire en ligne, consulté le )
- (en) Prusiner SB, « Shattuck lecture--neurodegenerative diseases and prions », The New England Journal of Medicine, vol. 344, no 20, , p. 1516–26 (PMID 11357156, DOI 10.1056/NEJM200105173442006)
- (en) Wen-Quan Zou et Pierluigi Gambetti, « From Microbes to Prions », Cell, vol. 121, no 2, , p. 155–157 (DOI 10.1016/j.cell.2005.04.002, lire en ligne, consulté le )
- (en) Jiyan Ma, « The Role of Cofactors in Prion Propagation and Infectivity », PLoS Pathogens, vol. 8, no 4, , e1002589 (ISSN 1553-7374, PMID 22511864, PMCID PMC3325206, DOI 10.1371/journal.ppat.1002589, lire en ligne, consulté le )
- (en) M. Meyer-Luehmann, « Exogenous Induction of Cerebral -Amyloidogenesis Is Governed by Agent and Host », Science, vol. 313, no 5794, , p. 1781–1784 (ISSN 0036-8075 et 1095-9203, DOI 10.1126/science.1131864, lire en ligne, consulté le )
- (en) Florence Clavaguera, Tristan Bolmont, R. Anthony Crowther et Dorothee Abramowski, « Transmission and spreading of tauopathy in transgenic mouse brain », Nature Cell Biology, vol. 11, no 7, , p. 909–913 (ISSN 1465-7392 et 1476-4679, PMID 19503072, PMCID PMC2726961, DOI 10.1038/ncb1901, lire en ligne, consulté le )
- R. Melki, « Les protéinopathies infectieuses de Parkinson et d’Alzheimer », sur Académie nationale de médecine, (consulté le )
- (en) P. Desplats, H.-J. Lee, E.-J. Bae et C. Patrick, « Inclusion formation and neuronal cell death through neuron-to-neuron transmission of -synuclein », Proceedings of the National Academy of Sciences, vol. 106, no 31, , p. 13010–13015 (ISSN 0027-8424 et 1091-6490, PMID 19651612, PMCID PMC2722313, DOI 10.1073/pnas.0903691106, lire en ligne, consulté le )
- (en) Christian Hansen, Elodie Angot, Ann-Louise Bergström et Jennifer A. Steiner, « α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells », Journal of Clinical Investigation, vol. 121, no 2, , p. 715–725 (ISSN 0021-9738, PMID 21245577, PMCID PMC3026723, DOI 10.1172/JCI43366, lire en ligne, consulté le )
- (en) Jeffrey H. Kordower, Hemraj B. Dodiya, Adam M. Kordower et Brian Terpstra, « Transfer of host-derived alpha synuclein to grafted dopaminergic neurons in rat », Neurobiology of Disease, vol. 43, no 3, , p. 552–557 (PMID 21600984, PMCID PMC3430516, DOI 10.1016/j.nbd.2011.05.001, lire en ligne, consulté le )
- (en) Jeffrey H Kordower, Yaping Chu, Robert A Hauser et Thomas B Freeman, « Lewy body–like pathology in long-term embryonic nigral transplants in Parkinson's disease », Nature Medicine, vol. 14, no 5, , p. 504–506 (ISSN 1078-8956 et 1546-170X, DOI 10.1038/nm1747, lire en ligne, consulté le )
- (en) Ruth Chia, M. Howard Tattum, Samantha Jones et John Collinge, « Superoxide Dismutase 1 and tgSOD1G93A Mouse Spinal Cord Seed Fibrils, Suggesting a Propagative Cell Death Mechanism in Amyotrophic Lateral Sclerosis », PLoS ONE, vol. 5, no 5, , e10627 (ISSN 1932-6203, PMID 20498711, PMCID PMC2869360, DOI 10.1371/journal.pone.0010627, lire en ligne, consulté le )
- (en) Christian Münch, John O’Brien et Anne Bertolotti, « Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells », Proceedings of the National Academy of Sciences, vol. 108, no 9, , p. 3548–3553 (ISSN 0027-8424 et 1091-6490, PMID 21321227, PMCID PMC3048161, DOI 10.1073/pnas.1017275108, lire en ligne, consulté le )
- (en) Pei-Hsien Ren, Jane E. Lauckner, Ioulia Kachirskaia et John E. Heuser, « Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates », Nature Cell Biology, vol. 11, no 2, , p. 219–225 (ISSN 1465-7392 et 1476-4679, PMID 19151706, PMCID PMC2757079, DOI 10.1038/ncb1830, lire en ligne, consulté le )
- (en) Margaret M.P. Pearce et Ron R. Kopito, « Prion-Like Characteristics of Polyglutamine-Containing Proteins », Cold Spring Harbor Perspectives in Medicine, vol. 8, no 2, , a024257 (ISSN 2157-1422, PMID 28096245, PMCID PMC5793740, DOI 10.1101/cshperspect.a024257, lire en ligne, consulté le )
- Yoshiaki Furukawa, Kumi Kaneko, Shoji Watanabe et Koji Yamanaka, « A Seeding Reaction Recapitulates Intracellular Formation of Sarkosyl-insoluble Transactivation Response Element (TAR) DNA-binding Protein-43 Inclusions », Journal of Biological Chemistry, vol. 286, no 21, , p. 18664–18672 (ISSN 0021-9258, PMID 21454603, PMCID PMC3099683, DOI 10.1074/jbc.m111.231209, lire en ligne, consulté le )
- (en) Carlo Condello, William F. DeGrado et Stanley B. Prusiner, « Prion biology: implications for Alzheimer's disease therapeutics », The Lancet Neurology, vol. 19, no 10, , p. 802–803 (ISSN 1474-4422 et 1474-4465, PMID 32949533, DOI 10.1016/S1474-4422(20)30274-X, lire en ligne, consulté le )
- (en) K. Lundmark, G. T. Westermark, A. Olsen et P. Westermark, « Protein fibrils in nature can enhance amyloid protein A amyloidosis in mice: Cross-seeding as a disease mechanism », Proceedings of the National Academy of Sciences, vol. 102, no 17, , p. 6098–6102 (ISSN 0027-8424 et 1091-6490, PMID 15829582, PMCID PMC1087940, DOI 10.1073/pnas.0501814102, lire en ligne, consulté le )
- (en) Xiaoying Fu, Tatsumi Korenaga, Li Fu et Yanming Xing, « Induction of AApoAII amyloidosis by various heterogeneous amyloid fibrils », FEBS Letters, vol. 563, nos 1-3, , p. 179–184 (DOI 10.1016/S0014-5793(04)00295-9, lire en ligne, consulté le )
- Tristan Bolmont, Florence Clavaguera, Melanie Meyer-Luehmann et Martin C. Herzig, « Induction of Tau Pathology by Intracerebral Infusion of Amyloid-β-Containing Brain Extract and by Amyloid-β Deposition in APP × Tau Transgenic Mice », The American Journal of Pathology, vol. 171, no 6, , p. 2012–2020 (ISSN 0002-9440, PMID 18055549, PMCID PMC2111123, DOI 10.2353/ajpath.2007.070403, lire en ligne, consulté le )
- (en) R. Morales, L. D. Estrada, R. Diaz-Espinoza et D. Morales-Scheihing, « Molecular Cross Talk between Misfolded Proteins in Animal Models of Alzheimer's and Prion Diseases », Journal of Neuroscience, vol. 30, no 13, , p. 4528–4535 (ISSN 0270-6474 et 1529-2401, PMID 20357103, PMCID PMC2859074, DOI 10.1523/JNEUROSCI.5924-09.2010, lire en ligne, consulté le )
- Clavaguera F, Hench J, Goedert M, Tolnay M, « Invited review: Prion-like transmission and spreading of tau pathology », Neuropathology and Applied Neurobiology, vol. 41, no 1, , p. 47–58 (PMID 25399729, DOI 10.1111/nan.12197)
- Jucker M, Walker LC, « Self-propagation of pathogenic protein aggregates in neurodegenerative diseases », Nature, vol. 501, no 7465, , p. 45–51 (PMID 24005412, PMCID 3963807, DOI 10.1038/nature12481)
- Mann DM, Snowden JS, « Frontotemporal lobar degeneration: Pathogenesis, pathology and pathways to phenotype », Brain Pathology, vol. 27, no 6, , p. 723–736 (PMID 28100023, DOI 10.1111/bpa.12486)
- (en) Nelson, Dickson, Trojanowski et Boyle, « Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report », Brain, vol. Online first, (DOI 10.1093/brain/awz099, lire en ligne)
- Revesz T, Ghiso J, Lashley T, Plant G, Rostagno A, Frangione B, Holton JL, « Cerebral amyloid angiopathies: a pathologic, biochemical, and genetic view », Journal of Neuropathology and Experimental Neurology, vol. 62, no 9, , p. 885–98 (PMID 14533778, DOI 10.1093/jnen/62.9.885
 )
) - Guo L, Salt TE, Luong V, Wood N, Cheung W, Maass A, Ferrari G, Russo-Marie F, Sillito AM, Cheetham ME, Moss SE, Fitzke FW, Cordeiro MF, « Targeting amyloid-beta in glaucoma treatment », Proceedings of the National Academy of Sciences of the United States of America, vol. 104, no 33, , p. 13444–9 (PMID 17684098, PMCID 1940230, DOI 10.1073/pnas.0703707104)
- SB Prusiner, Prion Biology and Diseases, Cold Spring Harbor, NY, Cold Spring Harbor Laboratory Press, , 2e éd. (ISBN 0-87969-693-1)
- Goedert M, Spillantini MG, Del Tredici K, Braak H, « 100 years of Lewy pathology », Nature Reviews. Neurology, vol. 9, no 1, , p. 13–24 (PMID 23183883, DOI 10.1038/nrneurol.2012.242)
- Grad LI, Fernando SM, Cashman NR, « From molecule to molecule and cell to cell: prion-like mechanisms in amyotrophic lateral sclerosis », Neurobiology of Disease, vol. 77, , p. 257–65 (PMID 25701498, DOI 10.1016/j.nbd.2015.02.009)
- Ludolph AC, Brettschneider J, Weishaupt JH, « Amyotrophic lateral sclerosis », Current Opinion in Neurology, vol. 25, no 5, , p. 530–5 (PMID 22918486, DOI 10.1097/WCO.0b013e328356d328)
- Orr HT, Zoghbi HY, « Trinucleotide repeat disorders », Annual Review of Neuroscience, vol. 30, no 1, , p. 575–621 (PMID 17417937, DOI 10.1146/annurev.neuro.29.051605.113042)
- Almeida B, Fernandes S, Abreu IA, Macedo-Ribeiro S, « Trinucleotide repeats: a structural perspective », Frontiers in Neurology, vol. 4, , p. 76 (PMID 23801983, PMCID 3687200, DOI 10.3389/fneur.2013.00076)
- Spinner NB, « CADASIL: Notch signaling defect or protein accumulation problem? », The Journal of Clinical Investigation, vol. 105, no 5, , p. 561–2 (PMID 10712425, PMCID 292459, DOI 10.1172/JCI9511)
- Quinlan RA, Brenner M, Goldman JE, Messing A, « GFAP and its role in Alexander disease », Experimental Cell Research, vol. 313, no 10, , p. 2077–87 (PMID 17498694, PMCID 2702672, DOI 10.1016/j.yexcr.2007.04.004)
- Ito D, Suzuki N, « Seipinopathy: a novel endoplasmic reticulum stress-associated disease », Brain, vol. 132, no Pt 1, , p. 8–15 (PMID 18790819, DOI 10.1093/brain/awn216 )
- Sipe JD, Benson MD, Buxbaum JN, Ikeda SI, Merlini G, Saraiva MJ, Westermark P, « Amyloid fibril proteins and amyloidosis: chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines », Amyloid, vol. 23, no 4, , p. 209–213 (PMID 27884064, DOI 10.1080/13506129.2016.1257986 )
- Lomas DA, Carrell RW, « Serpinopathies and the conformational dementias », Nature Reviews Genetics, vol. 3, no 10, , p. 759–68 (PMID 12360234, DOI 10.1038/nrg907)
- primary systemic amyloidosis
- Mukherjee A, Soto C, « Prion-Like Protein Aggregates and Type 2 Diabetes », Cold Spring Harbor Perspectives in Medicine, vol. 7, no 5, , a024315 (PMID 28159831, PMCID 5411686, DOI 10.1101/cshperspect.a024315)
- Ahmet Dogan, « Amyloidosis: Insights from Proteomics », Annual Review of Pathology, vol. 12, , p. 277–304 (ISSN 1553-4014, PMID 27959636, DOI 10.1146/annurev-pathol-052016-100200, lire en ligne, consulté le )
- (en) Roberto Scarpioni, Marco Ricardi, Vittorio Albertazzi et Sara De Amicis, « Dialysis-related amyloidosis: challenges and solutions », International Journal of Nephrology and Renovascular Disease, vol. Volume 9, , p. 319–328 (ISSN 1178-7058, PMID 27994478, PMCID PMC5153266, DOI 10.2147/IJNRD.S84784, lire en ligne, consulté le )
- Askanas V, Engel WK, « Inclusion-body myositis: a myodegenerative conformational disorder associated with Abeta, protein misfolding, and proteasome inhibition », Neurology, vol. 66, no 2 Suppl 1, , S39-48 (PMID 16432144, DOI 10.1212/01.wnl.0000192128.13875.1e)
- Ecroyd H, Carver JA, « Crystallin proteins and amyloid fibrils », Cellular and Molecular Life Sciences, vol. 66, no 1, , p. 62–81 (PMID 18810322, DOI 10.1007/s00018-008-8327-4)
- Surguchev A, Surguchov A, « Conformational diseases: looking into the eyes », Brain Research Bulletin, vol. 81, no 1, , p. 12–24 (PMID 19808079, DOI 10.1016/j.brainresbull.2009.09.015)
- Huilgol SC, Ramnarain N, Carrington P, Leigh IM, Black MM, « Cytokeratins in primary cutaneous amyloidosis », The Australasian Journal of Dermatology, vol. 39, no 2, , p. 81–5 (PMID 9611375, DOI 10.1111/j.1440-0960.1998.tb01253.x)
- « Lichen Amyloidosis: Background, Pathophysiology », (consulté le )
- Janig E, Stumptner C, Fuchsbichler A, Denk H, Zatloukal K, « Interaction of stress proteins with misfolded keratins », European Journal of Cell Biology, vol. 84, nos 2–3, , p. 329–39 (PMID 15819411, DOI 10.1016/j.ejcb.2004.12.018)
- (en) James P. Solomon, Lesley J. Page, William E. Balch et Jeffery W. Kelly, « Gelsolin amyloidosis: genetics, biochemistry, pathology and possible strategies for therapeutic intervention », Critical Reviews in Biochemistry and Molecular Biology, vol. 47, no 3, , p. 282–296 (ISSN 1040-9238 et 1549-7798, PMID 22360545, PMCID PMC3337338, DOI 10.3109/10409238.2012.661401, lire en ligne, consulté le )
- D'Souza A, Theis JD, Vrana JA, Dogan A, « Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration », Amyloid, vol. 21, no 2, , p. 71–5 (PMID 24446896, PMCID 4021035, DOI 10.3109/13506129.2013.876984)
- Meng X, Clews J, Kargas V, Wang X, Ford RC, « The cystic fibrosis transmembrane conductance regulator (CFTR) and its stability », Cellular and Molecular Life Sciences, vol. 74, no 1, , p. 23–38 (PMID 27734094, PMCID 5209436, DOI 10.1007/s00018-016-2386-8)
- Stuart MJ, Nagel RL, « Sickle-cell disease », Lancet, vol. 364, no 9442, , p. 1343–60 (PMID 15474138, DOI 10.1016/S0140-6736(04)17192-4)
- (en) Mark B. Pepys, « Amyloidosis », Annual Review of Medicine, vol. 57, no 1, , p. 223–241 (ISSN 0066-4219 et 1545-326X, DOI 10.1146/annurev.med.57.121304.131243, lire en ligne, consulté le )
- (en) D. M. Holtzman, J. C. Morris et A. M. Goate, « Alzheimer's Disease: The Challenge of the Second Century », Science Translational Medicine, vol. 3, no 77, , p. 77sr1–77sr1 (ISSN 1946-6234 et 1946-6242, PMID 21471435, PMCID PMC3130546, DOI 10.1126/scitranslmed.3002369, lire en ligne, consulté le )
- (en) M. B. Pepys, « Pathogenesis, diagnosis and treatment of systemic amyloidosis », Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences, vol. 356, no 1406, , p. 203–211 (ISSN 0962-8436 et 1471-2970, PMID 11260801, PMCID PMC1088426, DOI 10.1098/rstb.2000.0766, lire en ligne, consulté le )
- Lary C. Walker et Harry LeVine, « Proteopathy: the next therapeutic frontier? », Current Opinion in Investigational Drugs (London, England: 2000), vol. 3, no 5, , p. 782–787 (ISSN 1472-4472, PMID 12090553, lire en ligne, consulté le )
- (en) Anne K. Braczynski, Jörg B. Schulz et Jan-Philipp Bach, « Vaccination strategies in tauopathies and synucleinopathies », Journal of Neurochemistry, vol. 143, no 5, , p. 467–488 (DOI 10.1111/jnc.14207, lire en ligne, consulté le )
- William L. Klein, « Synaptotoxic Amyloid-β Oligomers: A Molecular Basis for the Cause, Diagnosis, and Treatment of Alzheimer's Disease? », Journal of Alzheimer's Disease, vol. 33, no s1, , S49–S65 (DOI 10.3233/JAD-2012-129039, lire en ligne, consulté le )
- (en) Talha Badar, Anita D'Souza et Parameswaran Hari, « Recent advances in understanding and treating immunoglobulin light chain amyloidosis », F1000Research, vol. 7, , p. 1348 (ISSN 2046-1402, PMID 30228867, PMCID PMC6117860, DOI 10.12688/f1000research.15353.1, lire en ligne, consulté le )
- (en) Andreia Carvalho, Ana Rocha et Luísa Lobato, « Liver transplantation in transthyretin amyloidosis: Issues and challenges: Liver Transplantation in Attr », Liver Transplantation, vol. 21, no 3, , p. 282–292 (DOI 10.1002/lt.24058, lire en ligne, consulté le )
- « Liver transplantation for hereditary transthyretin amyloidosis », Liver Transpl, vol. 6, no 3, , p. 263-276 (PMID 10827225, DOI 10.1053/lv.2000.6145)
- (en) Suhr OB, Larsson M, Ericzon BG, Wilczek HE et al., « Survival After Transplantation in Patients With Mutations Other Than Val30Met: Extracts From the FAP World Transplant Registry », Transplantation, vol. 100, no 2, , p. 373-381 (PMID 26656838, PMCID 4732012, DOI 10.1097/TP.0000000000001021).
- (en) Teresa Coelho, Giampaolo Merlini, Christine E. Bulawa et James A. Fleming, « Mechanism of Action and Clinical Application of Tafamidis in Hereditary Transthyretin Amyloidosis », Neurology and Therapy, vol. 5, no 1, , p. 1–25 (ISSN 2193-8253 et 2193-6536, PMID 26894299, PMCID PMC4919130, DOI 10.1007/s40120-016-0040-x, lire en ligne, consulté le )
- (en) Dongbo Yu, Hannah Pendergraff, Jing Liu et Holly B. Kordasiewicz, « Single-Stranded RNAs Use RNAi to Potently and Allele-Selectively Inhibit Mutant Huntingtin Expression », Cell, vol. 150, no 5, , p. 895–908 (PMID 22939619, PMCID PMC3444165, DOI 10.1016/j.cell.2012.08.002, lire en ligne, consulté le )
- (en) Mario Nuvolone et Giampaolo Merlini, « Emerging therapeutic targets currently under investigation for the treatment of systemic amyloidosis », Expert Opinion on Therapeutic Targets, vol. 21, no 12, , p. 1095–1110 (ISSN 1472-8222 et 1744-7631, DOI 10.1080/14728222.2017.1398235, lire en ligne, consulté le )
- (en) Nisha S. Joseph et Jonathan L. Kaufman, « Novel Approaches for the Management of AL Amyloidosis », Current Hematologic Malignancy Reports, vol. 13, no 3, , p. 212–219 (ISSN 1558-8211 et 1558-822X, DOI 10.1007/s11899-018-0450-1, lire en ligne, consulté le )