Rétinite pigmentaire
Les rétinites pigmentaires sont un ensemble de maladies génétiques de l'œil. Le nom de retinitis pigmentosa aurait été proposé en 1855 par le Néerlandais Franz Donders.
| Médicament | Voretigène néparvovec (en) |
|---|---|
| Spécialité | Ophtalmologie |
| CIM-10 | H35.5 |
|---|---|
| CIM-9 | 362.74 |
| OMIM | 268000 |
| DiseasesDB | 11429 |
| MedlinePlus | 001029 |
| MeSH | D012174 |
| GeneReviews | |
| Patient UK | Retinitis-pigmentosa |
![]() Mise en garde médicale
Mise en garde médicale
Cet ensemble est génétiquement hétérogène et implique les photorécepteurs (cônes et bâtonnets) et l'épithélium pigmentaire rétinien. Elles se manifestent d'abord par une perte de la vision nocturne suivie d'un rétrécissement du champ visuel. La perte de la vision centrale est tardive.
La rétinite pigmentaire peut être divisée en rétinite pigmentaire non syndromique, syndromique ou systémique atteignant d'autres organes. Cet article ne traite que des rétinites pigmentaires non syndromiques.
Incidence et prévalence
Une personne sur 4 000 est atteinte[1] - [2]. C'est la cause la plus fréquente de cécité chez la personne d'âge intermédiaire dans les pays développés.
Description
L'atteinte oculaire survient à des âges variables mais débute généralement tôt (10 à 20 ans) par une diminution de la vision nocturne (lésions préférentielles des bâtonnets, cellules de la rétine plus adaptées aux faibles luminosités), appelée héméralopie. De plus, le patient souffre également de photophobie (due à la détérioration de l'épithélium pigmentaire). Dans un second temps, le champ visuel se rétrécit, pouvant aboutir à une vision « en tunnel ». Les difficultés majeures (concernant les déplacements, la lecture, la conduite…) surviennent généralement à la trentaine. La cécité arrive avec la destruction des cônes souvent vers les 40 ans. L'atteinte est typiquement bilatérale, symétrique et diffuse.
Dans 30 % des cas, la rétinite est associée à des lésions d'autres organes formant une trentaine de syndromes. Le plus fréquent est le syndrome d'Usher associant une surdité. Le syndrome de Bardet-Biedl et la maladie de Refsum sont des syndromes plus rares.
Diagnostic
Le diagnostic se fait essentiellement par l'analyse du champ visuel retrouvant des scotomes périphériques (« trous » dans le champ de vision non central). Peuvent y être associés, selon le syndrome, un trouble de la vision des couleurs et/ou un trouble de la réfraction.
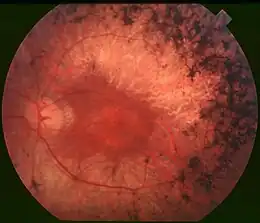
L'ophtalmoscope peut détecter une cataracte, typiquement sous-capsulaire, dans la moitié des cas. Le fond d'œil montre une hyperpigmentation de la périphérie de la rétine, témoin de la mort cellulaire. Un amincissement des vaisseaux rétiniens est aussi observé et, dans les stades avancés, le nerf optique présente une pâleur anormale.
L'électrorétinogramme permettrait un dépistage de la maladie avant que cette dernière se manifeste, mais présente très peu d'intérêt dans les stades évolués.
La tomographie en cohérence optique peut être utile[3].
Différents types
On distingue selon leur mode de transmission différents types de rétinite pigmentaire. Près de 45 types différents d'atteinte génétique ont été décrits, la plupart des gènes codant des protéines photo-sensibles des bâtonnets. Ces types correspondent à environ 60 % de rétinites pigmentaires héréditaires, le reste étant à gènes non identifiés[4].
| Transmission | Fréquence |
|---|---|
| Récessive | 5 à 20 % |
| Dominante | 15 à 25 % |
| Récessive liée à l'X | 5 à 15 % |
| Inconnue | 40 à 50 % |
| Digénique | Très rare |
Traitement et prise en charge
Maladie génétique, la rétinite est jusqu'aujourd'hui incurable.
Une supplémentation en vitamine A pourrait freiner l'évolution de la maladie[5]. De même, un régime enrichi en Oméga-3 retarderait l'apparition de la cécité[6].
Certains syndromes (rares) ont également un traitement spécifique efficace : un régime pauvre en acide phytanique peut stopper l'évolution de la maladie de Refsum.
Chez la souris, des bâtonnets d'individus sains ont été isolés et transplantés chez des individus atteints d'une forme de rétinite pigmentaire. Les bâtonnets greffés sécrètent une substance qui protège les cônes de la destruction. Cette technique devrait être applicable à l'homme.
La thérapie génique semble prometteuse[7].
De plus, une nouvelle voie de recherche s'oriente vers l'implantation de rétine artificielle, sous forme de matrice d'électrodes. La Food and Drug Administration (FDA) a autorisé le la première prothèse épirétinienne, appelée Argus II Retinal Prosthesis System destinée à traiter les patients atteints de rétinite pigmentaire évoluée[8].
Notes et références
- « Rétinite pigmentaire », sur Futura (consulté le ).
- (en) S Natarajan, « Retinitis pigmentosa: A brief overview », Indian Journal of Ophthalmology, vol. 59, no 5, , p. 343-346 (ISSN 0301-4738, PMID 21836337, DOI 10.4103/0301-4738.83608, lire en ligne, consulté le )
- Mitamura Y, Mitamura-Aizawa S, Nagasawa T, Katome T, Eguchi H, Naito T, Diagnostic imaging in patients with retinitis pigmentosa, J Med Invest, 2012;59:1–11
- Hartong DT, Berson EL, Dryja TP, Retinitis pigmentosa, Lancet, 2006;368:1795–1809
- (en) Berson EL, Rosner B, Sandberg MA et al. « A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa » Arch Ophthalmol. 1993; 111: 761–72.
- (en) Hoffman DR, Locke KG, Wheaton DH, Fish GE, Spencer R, Birch DG. « A randomized, placebo-controlled clinical trial of docosahexaenoic acid supplementation for X-linked retinitis pigmentosa » Am J Ophthalmol. 2004; 137: 704–18.
- Sahel JA, Roska B, Gene therapy for blindness, Annu Rev Neurosci, 2013;36:467–488
- FDA News release (consulté le 20 février 2013)
- (en) Roberta A Pagon, Stephen P Daiger, Retinitis Pigmentosa Overview In : GeneReviews at GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1997-2005. .
- (en) Dyonne T Hartong, Eliot L Berson, Thaddeus P Dryja « Retinitis pigmentosa » Lancet 2006; 368:1795–1809.
Liens externes
- Association SOS Rétinite, Association Nationale de Lutte contre la Cécité
- Association Rétine Active (Lutte contre la Rétinite Pigmentaire)
- Association I.R.R.P (Information et Recherche sur la Rétinite Pigmentaire)
- Association Retina France - vaincre les maladies de la vue
- (en) Publications scientifiques sur Medline