| épithéliales |
|---|
| type |
clinique |
mutations |
transmission |
siège |
lésions histologiques |
| dystrophie épithéliale microkystique de cogan ou map-dot-
fingerprint
(OMIM 121820) |
- la plus fréquente
- bilatérale
- début adolescence
- érosions récidivantes avec douleur au réveil
- opacités variables et labiles, grisâtres, centrales en points ou à bordure festonnée (en carte de géographie) |
gène TGFBI de la kérato-
épithéline |
sporadique ou autosomique dominant |
épithéliale |
- pseudokystes intra‑épithéliaux contenant des noyaux pycnotiques et des débris cytoplasmiques, colorés PAS+ (correspondant aux opacités en points)
- membrane basale multilamellaire aberrante et déformée en feuillets formant des invaginations en doigt de gant dans l’épithélium (correspondant aux opacités en carte de géographie), colorée violet en PAS et bleu au trichrome de Masson
- cellules épithéliales remaniées œdémateuses
- couche de Bowman normale |
| dystrophie juvénile de Meesmann (OMIM 122100) |
- bilatéral, symétrique
- début dans adolescence, possible petite enfance
- crises douloureuses par érosions épithéliales récidivantes
→ diagnostic possible sur simple pelage épithélial |
gène TGFBI des cyto-
kératines CK12 et CK3 |
autosomique dominant à pénétrance incomplète |
épithéliale |
- épithélium et MB irréguliers et épaissis et dyskératose/légère acanthose
- kystes PAS positifs intra‑épithéliaux avec matériel amorphe intrakystique
- c épithéliales avec halo clair périnucléaire
- mb basale épaissie multilamellaire
- couche de Bowman intacte |
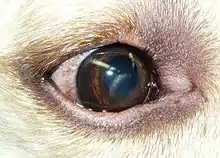
| dystrophie de Lisch (corneal band-shaped microkystic dystrophy) |
- début dans enfance
- opacités superficielles linéaires, radiaires, respectant le centre de la cornée, puis évolution vers atteinte centrale et BAV (trentaine) |
gène localisé en Xp22.3 |
récessive liée à l’X |
épithéliale |
- microkystes dans toute l’épaisseur de l’épithélium: vacuolisation cytoplasmique diffuse |
| érosion récidivante atraumatique (Franceschetti) |
- début = enfance ou adulte
- érosions récidivantes, photophobie intense, +/- BAV
- lésions centrales uni ou bilatérales avec bulles et kystes épithéliaux |
|
autosomique dominante |
épithéliale |
- micro‑kystes intra‑ épithéliaux
- œdème épithélial focal intra‑ et extra‑cellulaire
- défauts de cicatrisation de la membrane basale
- couche de Bowman endommagée avec pannus fibreux |
| dystrophie de Grayson-
Wilbrandt |
- début = enfant/ado
- érosions douloureuses + BAV
- bilatérale
- opacités centrales gris-blanc en mottes au niveau de la Bowman se prolongeant dans l’épithélium |
|
autosomique dominante |
épithéliale |
- dépôts PAS + au niveau de la membrane basale à extension intra‑ épithéliale
- dégénérescence des cellules basales
- absence focale de couche de Bowman |
| dystrophie de Fleisher-grüber (cornea verticillata) |
- début = sujet jeune
- lignes brunes arquées, en forme de comète centrée par un point
- en cornée inférieure
- angiokératomes cutanés diffus |
|
récessive liée à l’X |
épithéliale |
- dépôts de glycolipides dans les cellules épithéliales basales
- membrane basale irrégulière dédoublée
- couche de Bowman parfois atteinte |
| syndrome KID (kératite, ichtyose, surdité) |
- kératite (95%) + insuffisance limbique avec néovascularisation cornéenne + surdité de perception + érythrokératodermie + alopécie |
gène de la connexine 26
(protéine exprimée par les gap- junctions de épithélium cornéen, épiderme et annexes) |
autosomique dominant
sporadique ++ |
épithéliale |
- dyskératose de l’épithélium cornéen et conjonctival |
| stromales (+/- épithéliales) |
| type |
clinique |
mutations |
transmission |
siège |
lésions histologiques |
| dystrophie de Reis-Bücklers (ou dystrophie de la couche de Bowman de type I ou dystrophie géographique (OMIM 608470) |
- début = enfance
- érosions épithéliales récidivantes, avec BAV à 20–30 ans sur opacités bilatérales et symétriques, confluentes, à contours géographiques, situées dans le plan de la couche de Bowman au niveau de la cornée centrale, avec un aspect vitreux ou granuleux du stroma antérieur
- forme superficielle de dystrophie granulaire (aussi nommée dystrophie granulaire de type III) |
gène TGFBI de la kératoépithéline: R124L
Délétion F540 |
Autosomique dominante, pénétrance complète |
Couche de Bowman et stroma antérieur |
- disparitions localisées de la couche de Bowman remplacée par une couche de tissu conjonctif en feuillets
- avec dépôts granulaires de kératohyaline (fibrillaires) dans la couche de Bowman et dans le stroma antérieur, très éosinophiles et colorés en rouge intense par le trichrome de Masson
- épithélium sus-jacent épaissi irrégulier avec des prolongements dans le stroma antérieur
→ immuno réaction avec les anticorps anti-TGFBI |
| dystrophie de Thiel-Behnke (dystrophie de la couche de Bowman de type II, dystrophie en rayon de miel, OMIM 602082) |
- début = enfance
- érosions épithéliales récidivantes avec BAV possible mais tardive, sur opacités blanchâtres centrales sous‑épithéliales bilatérales en rayons de miel pathognomonique |
gène TGFBI KE: R555Q
Délétion F540 |
Autosomique dominante, pénétrance complète |
Couche de Bowman et stroma antérieur |
- destruction de la couche de Bowman remplacée par un tissu/pannus fibrocellulaire sous‑épithélial d’aspect ondulé (en dents de scie), coloré en bleu au trichrome de Masson
- épithélium constitué de cellules de taille et de forme irrégulières, formant des crêtes et des sillons dans le tissu fibreux sous-épithélial
- membrane basale dédoublée et épaissie qui disparaît à de nombreux endroits
- dépôts granulaires moins prononcés que la dystrophie de Reis-Bücklers et moins colorés rouge par le trichrome de Masson |
| dystrophie granulaire de type I (OMIM 121900) |
- début = petite enfance (2 ans), plutôt peu sévère
- érosions épithéliales récidivantes puis BAV progressive avec dépôts granuleux blanchâtres (en miettes de pain) au niveau des couches antérieure et moyenne du stroma central (lésions individuelles trop petites et nombreuses pour être facilement comptées, contrairement à la dystrophie granulaire de type II), séparés par des intervalles de cornée claire
→ 1°) forme diffuse superficielle (homozygote) = toute la surface de la cornée, dépôts au niveau de la Bowman et du stroma antérieur, aspect en verre de cathédrale
→ 2°) forme granuleuse pré-descemétique = petits dépôts cristallins blanchâtres postérieurs |
gène TGFBI KE: R555WD123H R124L
R124S
Arg555Trp
Dél T125E126 |
Autosomique dominante, pénétrance complète |
-Stroma antérieur et moyen + Bowman
-Stroma postérieur (forme pré-descemétique) |
- multiples dépôts granulaires (fibrillaires) irréguliers, bien circonscrits de materiau hyalin dans un stroma déformé, s’étendant de l'épithélium profond à la membrane de Descemet, colorés en rouge par le trichrome de Masson, + éosinophiles que le stroma normal et moins PAS positifs (ils se colorent aussi intensément par le luxol fast blue)
- épithélium irrégulier avec des zones de disparition de la couche de Bowman |
| dystrophie d’Avellino (dystrophie granulaire de type II, OMIM 607541) |
- début = enfance (ou forme homozygote précoce presque congénitale)
- lésions de type points blanchâtres superficiels de distribution en “feux d’artifice” puis évolution en lésions + volumineuses (en flocons de neige/anneaux) facilement dénombrables |
gène TGFBI KE: R124H |
Auto. dominante, pénétrance complète, expressivité variable |
Stroma |
- dépôts + diffus s'étendant de l'épithélium basal au stroma profond, colorés soit avec le trichrome de Masson (kératohyaline) soit avec le rouge Congo (amyloïde) → les dépôts antérieurs sont colorés avec les deux, tandis que les lignes de réseau + profondes ne se colorent qu'avec du rouge Congo
- pas de positivité rouge congo dans la forme homozygote |
| dystrophie grillagée de type I (OMIM 122200) |
- début = enfance (1° ou 2° décennie)
- bilatérale et symétrique avec érosions récidivantes puis BAV progressive sur dépôts punctiformes superficiels au centre de la cornée puis lignes cristallines irrégulières se dichotomisant en «crins de Florence» avec une coloration jaunâtre de la cornée, évolution vers extension profonde + périph
→ greffe vers 40 ans |
gène TGFBI KE: R124C
L569R
L518P
Double mutation A546D – P551Q |
Autosomique dominante, pénétrance complète |
Stroma et couche de Bowman |
- dépôts amyloïdes ovalaires/fusiformes mal délimités entre la MB et la couche de Bowman, et entre les lamelles du stroma, colorés en orange en rouge Congo, fluo après thioflavine T, biréfringents en lumière polarisée, positifs en PAS, positifs en cristal violet
- épithélium et couche de Bowman minces, irréguliers et partiellement détruits, et dont l’architecture lamellaire est déformée par les dépôts |
| dystrophie grillagée de type IIIA |
- début + tardif vers 40 ans, parfois après 70 ans
- dépôts + épais s’étendant de limbe à limbe, opacités nodulaires, puis voile diffus
→ greffe vers 40-70 ans |
gène TGFBI KE: P501T, A546T
N622K, V627S |
Autosomique dominante, pénétrance incomplète |
Stroma et couche de Bowman |
- dépôts amyloïdes (rouge Congo, lumière polarisée) |
| dystrophie grillagée intermédiaire de type I/IIIA |
- début = intermédiaire vers 20–40 ans, asymétrique |
|
AD, pénétrance complète |
|
|
| dystrophie grillagée profonde de type III |
- début vers 40–50 ans
- fortement asymétrique et lésions inaugurales dans le stroma postérieur puis progression vers le stroma antérieur (inverse des autres dystrophies grillagées) |
|
Autosomique dominante, pénétrance complète |
|
|
| dystrophie grillagée de type II (sd de Meretoja, OMIM 105120) |
- dépôts cornéens grillagés + neuropathie bilatérale et symétrique des nerfs crâniens et des nerfs périphériques + lichen amyloïde cutané |
Gène de la gelsoline |
Autosomique récessive |
Stroma et couche de Bowman |
- dépôts amyloïdes (rouge Congo, lumière polarisée), moins nombreux, plus discrets et plus allongés que la DG de type I |
| dystrophie maculaire (OMIM 217800) |
- début = adolescence mais possible avant 10 ans, plutôt sévère
- bilatérale et symétrique avec BAV et photophobie importantes + érosions
- dépôts sont mal limités, confluents, blanc sale dans toutes les couches du stroma, respectant une zone prélimbique, avec une brume entre les dépôts
- superficielle puis progresse vers le stroma post
- épaisseur et sensibilité cornéenne diminuées
→ possible atteinte des cellules endothéliales et cornea guttata
→ greffe avant 40 ans ++ |
gène CHST6 exon3 (anomalie du métabolisme du kératan sulfate): Q18X, P31S, R50C, S51L, G52D, S53L, L59P, V66L, P72S, Q71D, V76M, etc... |
Autosomique récessive |
Stroma,
mb de Descemet,
endothélium |
- dépôts de GAG intracellulaire dans les kératocytes du stroma et de l’endothélium (donnant un aspect discrètement vacuolisé), et extracellulaires dans le stroma, la membrane de Descemet et l’endothélium
- ces dépôts sont légèrement basophiles, PAS positifs et colorés par le bleu alcian et le fer colloïdal
Type I : absence de kératan sulfate dans le sérum
Type II : présence de kératan sulfate dans le sérum |
| dystrophie stromale cristalline de Schnyder (OMIM 121800) |
→ dépôts de cristaux de cholestérol et de phospholipides dans le stroma + dégénérescence fibrilles de collagène
- début = petite enfance
- BAV progressive + hypercholestérolémie familiale associée dans 2/3 des cas
- bilatérale et symétrique: haze (voile) et accumulation de cristaux irisés (absents dans la moitié des cas) dans la portion centrale du stroma
- atteinte centrale qui progresse avec un arc lipidique périphérique puis un haze en moyenne périphérie
→ greffe vers 50 ans |
gène UBIAD1: N102S, G177R, G98S, p.Asp240Asn, N102S |
Autosomique dominante |
Stroma central |
- dépôts anormaux de phospholipides (estérifiés et non estérifiés) intra et extracellulaires, et de cristaux de cholestérol (HDL-cholestérol) dans les cellules épithéliales basales, la couche de Bowman et le stroma
- mieux évaluée avec le Noir Soudan, le Oil Red O ou autres colorations lipidiques sur du tissu frais congelé |
| dystrophie en mosaïque de Vogt |
- début tardif (forme sénile) ou enfance
- pas de BAV
- opacités gris blanchâtre sous‑épithéliales polygonales en écailles (grains calcaires), avec lésions centrales bilatérales de la couche de Bowman |
|
Autosomique dominante |
Couche de Bowman et stroma antérieur |
- dépôts calcaires au niveau de la couche de Bowman et du stroma antérieur
|
| kératopathie en bandelette héréditaire |
- début = congénital, dans l’enfance ou chez l’adulte
- BAV tardive possible
- dépôts blanchâtres bilatéraux au niveau de la couche de Bowman en zone de fente palpébrale |
|
Autosomique dominante ou récessive liée à l’X |
Couche de Bowman et stroma antérieur |
- dépôts granuleux calciques (colorés par le rouge alizarine) et non calciques dans la couche de Bowman et le stroma antérieur |
| dystrophie gélatineuse en goutte (amyloïde primaire familiale de la cornée, OMIM 204870) |
→ = amylose cornéenne primitive (dépôts amyloïdes)
- début = petite enfance, Japon ++
- BAV progressive avec des érosions douloureuses dues à des nodules “muriforme” sous-épithéliaux, photophobie + larmoiement
→ forme typique en forme de mûre (mulberry type): dépôts nodulaires translucides, centraux, stroma antérieur
→ autres formes cliniques: opacités en bande, opacités stromales, forme kumquat- like, néovascularisation cornéenne fréquente |
gènes TACSTD2 et M1S1: RM1, Q118E, Q118X, C119S, S170X, V194E, Q207X |
Autosomique récessive |
Sous-
épithélial |
- dépôts amyloïdes (rouge Congo, lumière polarisée) entre l’épithélium et la couche de Bowman puis progressant dans le stroma antérieur
- possibles dépôts globulaires amorphes avec coloration positive pour le tissu élastique |
| dystrophie mouchetée de François (OMIM 121850) |
- bilatérale mais asymptomatique et de découverte fortuite lors d’un examen ophtalmo
- dépôts punctiformes blanchâtres ou annulaires diffus dans le stroma
→ ttt et greffe inutiles |
gène d’une protéine des phospho-
inositide kinase |
autosomique dominante |
Stroma |
- présence d’un sous-ensemble de kératocytes gonflés et vacuolisés anormaux contenant un matériel coloré par le bleu alcian ou le fer colloïdal (glycosaminoglycanes), et le noir Soudan ou Oil red O (lipides complexes) |
| dystrophie congénitale héréditaire stromale (Turpin) |
- bilatérale et symétrique
- cornée laiteuse (opacification stromale) présente dès la naissance (mais l’absence d’atteinte endothéliale la distingue de la dystrophie endothéliale héréditaire congénitale)
→ greffe précoce car affection amblyogène |
gène de la décorine |
autosomique dominante, pénétrance complète |
Stroma |
- l’architecture peut être d’apparence normale mais on recherche une dissociation fibrillaire des couches stromales: lacunes remplies de filaments et des dépôts colorés par le bleu alcian |
| cornea farinata (dystrophie pré-descemétique) |
- début entre 30 et 60 ans
- saupoudrage blanchâtre très fin et diffus, en avant de la membrane de Descemet |
|
autosomique dominante |
Stroma postérieur |
- dépôts PAS positifs intracytoplasmiques dans les fibroblastes du stroma postérieur
- membrane de Descemet normale |
| dystrophie cristalline marginale de Bietti (OMIM 210370) |
- rétinopathie pigmentaire avec microcristaux dans toutes les couches de la rétine
- atteinte cornéenne asymptomatique: dépôts cristallins jaunâtres dans le stroma antérieur de la cornée périphérique, sous la couche de Bowman |
gène CYP4V2 (code protéine de la famille cytochrome P450) |
autosomique récessive |
Stroma antérieur |
- cristaux biréfringents de 10 à 20 μm de diamètre dans le stroma cornéen, les kératocytes, la rétine, les fibroblastes conjonctivaux et cutanés et dans les lymphocytes circulants |
| endothéliales |
| type |
clinique |
mutations |
transmission |
siège |
lésions histologiques |
| dystrophie endothéliale héréditaire congénitale (OMIM 121700, 217700) |
- cornée trouble/laiteuse dès la naissance avec atteinte endothéliale,
→ type 1: début la 1ère année de vie, asymétrique, photophobie et larmoiement sans nystagmus, progression sur 1 à 10 ans
→ type 2: non progressive avec nystagmus, amblyopie, HIO, kératopathie
→ greffe dès le diagnostic |
|
type 1: récessive
type 2: dominante |
endothélial |
- épaississement diffus de la membrane de Descemet
- cellules endothéliales clairsemées et atrophiques
- mieux évalué en PAS et trichrome de Masson
|
| dystrophie postérieure polymorphe
(OMIM 122000) |
- rarement congénitale
- opacification œdémateuse et kératopathie secondaire en forme de bande sous-épithéliale
- lésions endothéliales vésiculaires et alvéolaires isolées ou groupées |
au - 3 gènes différents |
autosomique dominante, pénétrance incomplète |
endothélial |
- endothélium fin atrophique remplacé focalement par des agrégats superposés ou multi-couches de cellules épithéliales en fuseau
- membrane Descemet épaissie avec des couches anormales de collagène, et des excroissances focales nodulaires et fusiformes
- mieux évalué en PAS et trichrome de Masson |
| dystrophie endothéliale de Fuchs (OMIM 136800) |
- forme classique début vers 40 ans, forme + précoce possible (1° décennie)
- vision altérée le matin s'améliorant spontanément au cours de la journée, avec ou sans érosions par éclatements de bulles épithéliales
- la décompensation endothéliale provoque un œdème stromal évoluant vers une kératopathie bulleuse entraînant des cicatrices fibreuses |
|
autosomique dominante |
endothélial |
- épaississement laminaire diffus de la membrane de Descemet, parsemée d’excroissances hyalines en forme de goutte/champignon qui peuvent faire saillie dans la chambre antérieure
- associé à des cellules endothéliales de densité diminuée, devenant hypertrophiées/aplaties pour combler les lacunes
- tissu fibreux sous-épithélial (pannus) fréquent dans les cas avancés |
| dystrophie endothéliale
liée à l’X |
- progressive chez les hommes mais pas les femmes
- chez l’homme: opacification laiteuse congénitale de la brume diffuse au verre dépoli, amblyopie, nystagmus
- chez la femme: possibles modifications endothéliales de type cratère lunaire asymptomatiques |
|
lié à l’X |
endothélial |
- amincissement irrégulier de l'épithélium et de la couche de Bowman
- disposition irrégulière des lamelles de collagène du stroma antérieur
- mb de Descemet irrégulièrement épaissie avec de petites excavations
- perte de cellules endothéliales |
 ;
;