Protéostase
La protéostase, terme provenant de protéine et homéostasie, est le concept qu'il existe des voies biologiques concurrentes et intégrées dans les cellules qui contrôlent la biogenèse, le repliement, le trafic et la dégradation des protéines présentes à l'intérieur et à l'extérieur de la cellule[1] - [2]. Le concept de maintien de la protéostase est essentiel pour comprendre la cause de maladies associées à un repliement des protéines excessif et à une dégradation conduisant à des phénotypes entraînant une perte de fonction[3], ainsi qu'aux troubles dégénératifs associés à l'agrégation[4]. Par conséquent, l'adaptation de la protéostase devrait permettre sa restauration une fois que sa perte a entraîné une pathologie. La protéostase cellulaire est essentielle pour assurer un développement réussi, un vieillissement en bonne santé, une résistance aux stress environnementaux et pour minimiser les perturbations de l'homéostasie par des agents pathogènes tels que les virus[2]. Les mécanismes par lesquels la protéostase est assurée comprennent la traduction régulée des protéines, le repliement assisté des protéines chaperons et les voies de dégradation des protéines. Il est essentiel d'ajuster chacun de ces mécanismes à la demande de protéines pour que toutes les fonctions cellulaires reposent sur un protéome correctement replié.
Mécanismes de la protéostase
Les rôles du ribosome dans la protéostase
L'un des premiers points de régulation de la protéostase est la traduction. Ceci est accompli via la structure du ribosome, un complexe essentiel à la traduction. Ces deux caractéristiques déterminent le repliement de la protéine et influencent ses futures interactions. La synthèse d'une nouvelle chaîne peptidique utilisant le ribosome est très lente et le ribosome peut même être bloqué lorsqu'il rencontre un codon rare, un codon trouvé à de faibles concentrations dans la cellule[5]. Ces pauses offrent la possibilité à un domaine protéique individuel de disposer du temps nécessaire pour se replier avant la production des domaines suivants. Cela facilite le repliement correct des protéines multi-domaines[5]. La chaîne peptidique nouvellement synthétisée quitte le ribosome dans l'environnement cellulaire par le canal de sortie étroit du ribosome (largeur : 10Å à 20Å, longueur : 80Å)[5]. En raison du manque d'espace dans le canal de sortie, la chaîne naissante forme déjà des structures secondaires et tertiaires limitées. Par exemple, une hélice alpha est une propriété structurelle de ce type qui est communément induite dans ce canal de sortie[6]. Dans le même temps, le canal de sortie empêche également le repliement prématuré en empêchant les interactions à grande échelle au sein de la chaîne peptidique, ce qui nécessiterait plus d'espace.
Molécules chaperonnes et maintien post-traductionnel de la protéostase
Afin de maintenir l'homéostasie protéique après la traduction, la cellule utilise des protéines chaperons, parfois des chaperonines, qui facilitent l'assemblage ou le désassemblage des protéines[7]. Elles reconnaissent les segments exposés d'acides aminés hydrophobes dans la chaîne peptidique naissante et travaillent ensuite à promouvoir la formation appropriée d'interactions non-covalentes conduisant à l'état replié souhaité[7]. Les chaperons commencent à aider au repliement des protéines dès qu'une chaîne naissante de plus de 60 acides aminés émerge du canal de sortie du ribosome[8]. L'un des chaperons de liaison aux ribosomes les plus étudiés est le facteur déclencheur. Ce facteur permet de stabiliser le peptide, favorise son repliement, empêche son agrégation et favorise le repliement de substrats modèles dénaturés[9]. Le facteur déclencheur agit non seulement directement sur le repliement correct de la protéine, mais recrute également d'autres chaperons dans le ribosome, tels que Hsp70. Hsp70 entoure une chaîne peptidique non pliée, empêchant ainsi l'agrégation et favorisant le repliement[7] - [8].
Les chaperonines sont une classe spéciale de chaperons qui favorisent le repliement de l'état natif en encapsulant de manière cyclique la chaîne peptidique[8]. Les chaperonines sont divisés en deux groupes. Les chaperonines du groupe 1 se trouvent couramment dans les bactéries, les chloroplastes et les mitochondries. Les chaperonines du groupe 2 se retrouvent à la fois dans le cytosol des cellules eucaryotes et dans les archées[10]. Les chaperonines du groupe 2 contiennent également un composant hélicoïdal supplémentaire qui agit comme un couvercle pour la chambre protéique cylindrique, contrairement au groupe 1 qui repose plutôt sur un co-chaperon supplémentaire servant de couvercle. Toutes les chaperonines présentent deux états (ouvert et fermé), entre lesquels ils peuvent faire un cycle. Ce processus cyclique est important lors du repliement d'une chaîne polypeptidique individuelle, car il permet d'éviter des interactions indésirables ainsi que d'empêcher le peptide d'entrer dans des états piégés cinétiquement[10].
Réguler la protéostase par dégradation des protéines
Le troisième composant du réseau de protéostase est la machinerie de dégradation des protéines. La dégradation des protéines se produit dans la protéostase lorsque les signaux cellulaires indiquent la nécessité de réduire les niveaux globaux de protéines cellulaires. Les effets de la dégradation des protéines peuvent être locaux, la cellule ne subissant que les effets de la perte de la protéine dégradée elle-même ou généralisés, tout le paysage protéique étant modifié en raison de la perte des interactions des autres protéines avec la protéine dégradée[6]. Les substrats multiples sont des cibles pour la dégradation protéostatique. Ces substrats dégradables comprennent des fragments de protéines non fonctionnels produits à partir du blocage du ribosome au cours de la traduction, des protéines mal repliées ou non pliées, des protéines agrégées et des protéines qui ne sont plus nécessaires pour assurer la fonction cellulaire. Plusieurs voies différentes existent pour mener à bien ces processus de dégradation. Lorsqu'il est déterminé que les protéines sont dépliées ou mal repliées, elles sont généralement dégradées via la réponse protéique dépliée (UPR pour unfolded protein response) ou la dégradation des protéines associée au réticulum endoplasmique (ERAD pour endoplasmic-reticulum-associated protein degradation). Les substrats dépliés, mal repliés ou qui ne sont plus nécessaires à la fonction cellulaire peuvent également être marqués à l'ubiquitine pour être dégradés par des protéases dépendant de l'ATP, tels que le protéasome chez les eucaryotes ou ClpXP chez les procaryotes. L'autophagie, ou auto-digestion, le ciblage lysosomal et la phagocytose (digestion de déchets par d'autres cellules) peuvent également être utilisés comme mécanismes de dégradation protéostatique[6].
Événements de signalisation dans la protéostase
Le mauvais repliement des protéines est détecté par des mécanismes spécifiques au compartiment cellulaire dans lequel elles se produisent. Des mécanismes de surveillance distincts qui répondent aux protéines non pliées ont été caractérisés dans le cytoplasme, le RE et les mitochondries. Cette réponse agit localement dans la cellule de manière autonome mais peut également s'étendre à la signalisation intercellulaire pour protéger l'organisme des stress protéotoxiques anticipés.
Réponse au stress cellulaire autonome
Les voies de réponse au stress cellulaire détectent et atténuent le stress protéotoxique qui est déclenché par des déséquilibres de la protéostase. La régulation cellulaire autonome résulte de la détection directe de protéines mal repliées ou de l'inhibition de l'activation de la voie en séquestrant des composants activateurs consécutifs au choc thermique. Les réponses cellulaires à cette signalisation de stress comprennent l'activation de la transcription de l'expression des chaperonnes, l'efficacité accrue du trafic de protéines, la dégradation des protéines et la réduction de la traduction.
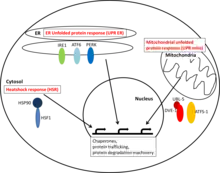
Réponse au choc thermique cytosolique
La HSR cytosolique est principalement médiée par la famille de facteurs de transcription HSF (famille du choc thermique ou heat shock family). HSF est liée de manière constitutive à Hsp90. Lors d'un stimulus protéotoxique, Hsp90 est recruté hors du HSF, lequel peut ensuite se lier aux éléments de réponse à la chaleur dans l'ADN et réguler positivement l'expression génique de protéines impliquées dans le maintien de la protéostase.
Réponse du RE en protéines dépliées
La réponse protéique non pliée dans le réticulum endoplasmique (RE) est activée par des déséquilibres de protéines non pliées à l'intérieur du RE et par l'homéostasie des protéines médiatrices. Différents «détecteurs» - tels que IRE1, ATF6 et PERK - peuvent reconnaître des protéines mal repliées dans le RE et médier des réponses transcriptionnelles qui aident à atténuer les effets du stress du RE.
Réponse mitochondriale en protéines non pliées
La réponse protéique non pliée mitochondriale détecte les déséquilibres de la stœchiométrie protéique des protéines mitochondriales et des protéines mal repliées. L'expression des molécules chaperonnes mitochondriales est régulée positivement par l'activation des facteurs de transcription ATF-1 et / ou DVE-1 avec UBL-5.
Signalisation systémique du stress
Les réponses au stress peuvent également être déclenchées de manière non cellulaire autonome par une communication intercellulaire. Le stress détecté dans un tissu pourrait ainsi être communiqué à d'autres tissus afin de protéger le protéome de l'organisme ou de réguler la protéostase de manière systémique. Une activation cellulaire non autonome peut se produire pour les trois réponses au stress.
Des travaux sur l'organisme modèle C. elegans ont montré que les neurones jouent un rôle dans cette communication intercellulaire de la HSR cytosolique. Le stress induit dans les neurones du ver peut à long terme protéger d'autres tissus tels que les cellules musculaires et intestinales contre la protéotoxicité chronique. De même, les ERP et UPR mitochondriales dans les neurones sont relayés aux cellules intestinales. Ces réponses systémiques ont été impliquées dans la médiation non seulement de la protéostase systémique, mais ont également une influence sur le vieillissement de l'organisme[11]
Maladies de la protéostase
Protéostase et maladies du repliement des protéines
Un dysfonctionnement de la protéostase peut résulter d'erreurs ou d'une mauvaise régulation du repliement des protéines. Les exemples classiques sont les mutations faux-sens et les délétions qui modifient les paramètres thermodynamiques et cinétiques pour le processus de repliement des protéines[1]. Ces mutations sont souvent héréditaires et leur sévérité phénotypique varie, allant de l'absence d'effet perceptible à la létalité embryonnaire. La maladie se développe lorsque ces mutations rendent une protéine nettement plus susceptible au repliement, à l'agrégation et à la dégradation. Si ces effets ne modifient que la protéine mutée, les conséquences négatives ne seront qu'une perte de fonction locale. Cependant, si ces mutations se produisent dans un chaperon ou une protéine qui interagit avec de nombreuses autres protéines, des altérations globales spectaculaires de l'ensemble de la protéostase se produiront. Des exemples de maladies résultant de modifications protéostatiques dues à des erreurs de repliement de protéines incluent la fibrose kystique, la maladie de Huntington, la maladie d'Alzheimer, les troubles de stockage lysosomal[12].
Le rôle des systèmes modèles dans l'élucidation des maladies à repliement protéique
Les systèmes de modèles animaux de petite taille ont joué et continuent de jouer un rôle déterminant dans l'identification de mécanismes fonctionnels préservant la protéostase. Les systèmes modèles de diverses protéines pathogènes à prédisposition au repliement ont déjà révélé de nombreux modificateurs chaperons et co-chaperons de protéotoxicité[13].
Protéostase et cancer
La division cellulaire non régulée qui marque le développement du cancer nécessite une synthèse protéique accrue pour la fonction et la survie des cellules cancéreuses. Cette augmentation de la synthèse protéique est généralement observée dans les protéines qui modulent le métabolisme cellulaire et les processus de croissance. Les cellules cancéreuses sont parfois sensibles aux médicaments qui inhibent les chaperonnes et perturbent la protéostase, tels que les inhibiteurs de Hsp90 ou les inhibiteurs du protéasome[1]. De plus, les cellules cancéreuses ont tendance à produire des protéines mal repliées, qui sont principalement éliminées par protéolyse[14]. Les inhibiteurs de la protéolyse permettent l'accumulation de protéines mal repliés, ainsi que les protéines de signalisation de l'apoptose dans les cellules cancéreuses[15] - [16]. Cela peut modifier la sensibilité des cellules cancéreuses aux médicaments anti-néoplasiques. les cellules cancéreuses meurent à une concentration plus faible du médicament ou survivent en fonction du type de protéines qui s'accumulent et de la fonction de ces protéines[17]. Le bortézomib, inhibiteur du protéasome, a été le premier médicament de ce type à être approuvé pour le traitement du myélome multiple[18].
Protéostase et obésité
Une caractéristique des réseaux protéostatiques cellulaires est leur capacité à s'adapter au stress via la régulation des protéines. Les maladies métaboliques, telles que celles associées à l'obésité, altèrent la capacité des réseaux de protéostase cellulaire à s'adapter au stress, souvent avec des effets néfastes sur la santé. Par exemple, lorsque la production d'insuline dépasse la capacité de sécrétion d'insuline de la cellule, un effondrement protéostatique se produit et la production de chaperonnes est gravement altérée. Cette perturbation entraîne les symptômes de la maladie chez les diabétiques[1].
Protéostase et vieillissement
Au fil du temps, le réseau de protéostase est chargé de protéines modifiées par des espèces réactives de l'oxygène et des métabolites qui induisent des dommages oxydatifs[1]. Ces sous-produits peuvent réagir avec les protéines cellulaires pour provoquer un mauvais repliement et une agrégation (en particulier dans les cellules ne se divisant pas comme les neurones). Ce risque est particulièrement élevé pour les protéines intrinsèquement désordonnées. Chez C. elegans, il a été démontré que la voie de l'IGFR-1 protège contre ces agrégats nuisibles, et des travaux expérimentaux ont suggéré que la régulation à la hausse du récepteur du facteur de croissance de l'insuline 1 (IGFR-1) pourrait stabiliser le réseau protéostatique et prévenir les effets néfastes du vieillissement[1]. L'expression du chaperome, à savoir l'ensemble des chaperons et des co-chaperons qui interagissent dans un réseau complexe de machines de repliement moléculaire pour réguler la fonction du protéome, est radicalement réprimée dans les cerveaux humains vieillissants et dans le cerveau des patients atteints de maladies neurodégénératives. Des analyses fonctionnelles sur C. elegans et des cellules humaines ont identifié un sous-réseau conservé du chaperome de 16 gènes chaperons, correspondant à 28 orthologues humains comme protection de protéostase dans le vieillissement et les maladies neurodégénératives du vieillissement[19].
Intervention pharmacologique dans la protéostase
Deux approches principales ont été utilisées pour le développement thérapeutique ciblant le réseau protéostatique : les chaperons pharmacologiques et les régulateurs de la protéostase. Le principe qui sous-tend la conception de chaperons pharmacologiques pour une intervention dans les maladies liées à la protéostase consiste à concevoir de petites molécules stabilisant des protéines présentant une stabilité limite. Auparavant, cette approche était utilisée pour cibler et stabiliser les récepteurs couplés à la protéine G, les récepteurs des neurotransmetteurs, les glycosidases, les protéines de stockage lysosomales et la protéine CFTR mutante responsable de la fibrose kystique et de la transthyrétine, lesquels peuvent être défectueux et s'agréger, conduisant à l'amyloïdose[1]. Vertex Pharmaceuticals et Pfizer vendent des chaperons pharmacologiques approuvés par les organismes de réglementation pour améliorer, respectivement, la fibrose kystique et les amyloïdoses transthyrétiniques[20]. Amicus vend un chaperon pharmacologique approuvé par un organisme de réglementation pour la maladie de Fabry - une maladie de stockage lysosomal.
Le principe des régulateurs de protéostase est différent, ces molécules altèrent la biologie du repliement et / ou de la dégradation des protéines en modifiant la stœchiométrie des composants du réseau de protéostase dans un compartiment sous-cellulaire donné. Par exemple, certains régulateurs de protéostase initient une signalisation sensible au stress, telle que la réponse protéique non pliée, qui reprogramme de manière transcriptionnelle le réseau de protéostase du réticulum endoplasmique[21]. Il a été suggéré que cette approche pourrait même être appliquée à titre prophylactique, telle que la régulation à la hausse de certaines voies de protection avant de subir un stress cellulaire grave anticipé. Un mécanisme théorique de cette approche comprend la régulation positive de la réponse au choc thermique pour empêcher les protéines de se dégrader lors du stress cellulaire[1].
Notes et références
- Powers, E.T., Morimoto, R.I., Dillin, A. et Kelly, J.W., « Biological and Chemical Approaches to Diseases of Proteostasis Deficiency », Annu. Rev. Biochem., vol. 78, , p. 959–91 (PMID 19298183, DOI 10.1146/annurev.biochem.052308.114844)
- « Adapting proteostasis for disease intervention », Science, vol. 319, no 5865, , p. 916–919 (PMID 18276881, DOI 10.1126/science.1141448)
- Mu, T-W., Ong, D.S.T., Wang, Y-J et Balch, W. E., « Chemical and Biological Approaches Synergize to Ameliorate Protein-Folding Diseases », Cell, vol. 134, no 5, , p. 769–781 (PMID 18775310, PMCID 2650088, DOI 10.1016/j.cell.2008.06.037)
- Cohen, E., Paulsson, J. F., Blinder, P., Burstyn-Cohen, T., Du, D., Estepa, G., Adame, A., Pham, H. M., Holzenberger, M., Kelly, J. W., Masliah, E. & Dillin, A., « Reduced IGF-1 signaling delays age-associated proteotoxicity in mice », Cell, vol. 139, no 6, , p. 1157–69 (PMID 20005808, PMCID 3017511, DOI 10.1016/j.cell.2009.11.014)
- Cavagnero, S. et Fedyukina, D. V., « Protein Folding at the Exit Tunnel », Annual Review of Biophysics, vol. 40, , p. 337–359 (PMID 21370971, PMCID 5807062, DOI 10.1146/annurev-biophys-042910-155338)
- Bustamante, C. J., et.al., « Mechanisms of Cellular Proteostasis: Insights from Single-Molecule Approaches », Annual Review of Biophysics, vol. 43, , p. 119–140 (PMID 24895851, PMCID 4620553, DOI 10.1146/annurev-biophys-051013-022811)
- Ye, K., et.al., « Molecular Chaperone Functions in Protein Folding and Proteostasis », Annual Review of Biophysics, vol. 82, , p. 323–355 (PMID 23746257, DOI 10.1146/annurev-biochem-060208-092442)
- Vabulas, M. R. et. al., « Protein Folding in the Cytoplasm and the Heat Shock Response », Cold Spring Harb Perspect Biol, vol. 2, no 12, , a004390 (PMID 21123396, PMCID 2982175, DOI 10.1101/cshperspect.a004390)
- Hoffman, A., « Structure and function of the molecular chaperone Trigger Factor », Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, vol. 1803, no 6, , p. 650–661 (PMID 20132842, DOI 10.1016/j.bbamcr.2010.01.017)
- Yébenes, H., et. al., « Chaperonins: two rings for folding », Trends Biochem Sci, vol. 36, no 8, , p. 424–432 (PMID 21723731, DOI 10.1016/j.tibs.2011.05.003)
- « Systemic stress signalling: understanding the cell non-autonomous control of proteostasis », Nature Reviews Molecular Cell Biology, vol. 15, no 3, , p. 506–14 (PMID 24556842, PMCID 5922984, DOI 10.1038/nrm3752)
- « Proteostasis impairment in protein-misfolding and aggregation diseases », Trends Cell Biol., vol. 24, no 9, , p. 211–217 (PMID 24946960, DOI 10.1016/j.tcb.2014.05.003)
- « Model systems of protein-misfolding diseases reveal chaperone modifiers of proteotoxicity », Dis Model Mech., vol. 9, no 8, , p. 823–838 (PMID 27491084, PMCID 5007983, DOI 10.1242/dmm.024703)
- Cohen-Kaplan V, Livneh I, Avni N, Cohen-Rosenzweig C, Ciechanover A, « The ubiquitin-proteasome system and autophagy: Coordinated and independent activities », Int J Biochem Cell Biol, vol. 79, , p. 403–418 (PMID 27448843, DOI 10.1016/j.biocel.2016.07.019)
- Moschovi M, Critselis E, Cen O, Adamaki M, Lambrou GI, Chrousos GP, Vlahopoulos S, « Drugs acting on homeostasis: challenging cancer cell adaptation », Expert Rev Anticancer Ther, vol. 15, no 12, , p. 1405–17 (PMID 26523494, DOI 10.1586/14737140.2015.1095095)
- Sionov RV, Vlahopoulos SA, Granot Z, « Regulation of Bim in Health and Disease », Oncotarget, vol. 6, no 27, , p. 23058–134 (PMID 26405162, PMCID 4695108, DOI 10.18632/oncotarget.5492)
- Lambrou GI, Papadimitriou L, Chrousos GP, Vlahopoulos SA, « Glucocorticoid and proteasome inhibitor impact on the leukemic lymphoblast: multiple, diverse signals converging on a few key downstream regulators », Mol. Cell. Endocrinol., vol. 351, no 2, , p. 142–51 (PMID 22273806, DOI 10.1016/j.mce.2012.01.003)
- Adams J, « Proteasome inhibition in cancer: development of PS-341 », Semin Oncol, vol. 28, no 6, , p. 613–9 (PMID 11740819, DOI 10.1016/s0093-7754(01)90034-x)
- « A conserved chaperome sub-network safeguards protein homeostasis in aging and neurodegenerative disease », Cell Rep., vol. 9, no 3, , p. 1135–1150 (PMID 25437566, PMCID 4255334, DOI 10.1016/j.celrep.2014.09.042)
- Bulawa C.E., Connelly S., DeVit M., Wang L. Weigel, Fleming J. Packman, Powers E.T., Wiseman R.L., Foss T.R., Wilson I.A., Kelly J.W., Labaudiniere R., « Tafamidis, A Potent and Selective Transthyretin Kinetic Stabilizer That Inhibits the Amyloid Cascade », Proc. Natl. Acad. Sci., vol. 109, no 24, , p. 9629–9634 (PMID 22645360, PMCID 3386102, DOI 10.1073/pnas.1121005109)
- Plate L., Cooley C.B., Chen J.J., Paxman R.J., Gallagher C.M., Madoux F., Genereux J.C., Dobbs W., Garza D., Spicer T.P., Scampavia L., Brown S.J., Rosen H., Powers E.T., Walter P., Hodder P., Wiseman R.L., Kelly J.W., « Small Molecule Proteostasis Regulators that Reprogram the ER to Reduce Extracellular Protein Aggregation », eLife, vol. 5, , p. 15550 (DOI 10.7554/elife.15550)