Structure secondaire
En biochimie et en biologie structurale, la structure secondaire se rapporte uniquement à la description de la structure tridimensionnelle localement adoptée par certains segments de molécules biologiques (molécules définies comme étant des biopolymères, comme c’est le cas pour les protéines et les acides nucléiques (ADN/ARN)). On parlera ensuite de structure tertiaire pour décrire la position relative (dans l'espace) de ces différents éléments de structure secondaire les uns par rapport aux autres.
La structure secondaire n'est définie que par les liaisons hydrogène à l'intérieur du biopolymère, telle qu'on peut les observer sur des structures à la résolution atomique. Dans les protéines, la structure secondaire est définie par des arrangements de liaisons hydrogène entre les groupements amide et carbonyle du squelette peptidique (les liaisons hydrogène impliquant les chaînes latérales des acides aminés ne sont pas prises en considération dans la structure secondaire). Dans les acides nucléiques, la structure secondaire est définie par les liaisons hydrogène entre les bases nucléiques.
L'arrangement des liaisons hydrogène est cependant corrélé à d'autres paramètres structuraux, ce qui a conduit à des définitions moins strictes de la structure secondaire. Par exemple, dans les hélices de protéines, le squelette peptidique adopte des angles dièdres qui sont localisés dans une zone spécifique du diagramme de Ramachandran. En conséquence, un segment d'acides aminés dont la conformation correspond à de tels angles dièdres est souvent qualifié d'hélice, qu'il présente ou non l'arrangement canonique de liaisons hydrogène. Plusieurs autres définitions moins formelles ont été proposées, souvent en s'inspirant de concepts issus de géométrie différentielle, comme la courbure ou la torsion. Enfin, les biologistes structuraux, lorsqu'ils résolvent une nouvelle structure, déterminent parfois la structure secondaire « à l'œil », de manière qualitative, et font figurer cette information dans le ficher de structure déposé à la protein data bank (PDB).
Le contenu brut en structures secondaires d'un biopolymère (par exemple, « cette protéine est composée à 40 % d'hélice α et à 20 % de feuillet β ») peut souvent être estimé de manière spectroscopique. Pour les protéines, la méthode la plus courante est le dichroïsme circulaire dans l'UV lointain (170-250 nm). Un double minima prononcé à 208 nm et 222 nm indique une structure en hélices α, tandis qu'un minimum simple à 204 nm ou 217 nm indique respectivement une structure en pelote aléatoire ou en feuillets β. La spectroscopie infrarouge, moins utilisée, permet de détecter des différences de paramètres vibrationnels dans les liaisons amides, qui résultent de la formation des liaisons hydrogène. Enfin, le contenu en structures secondaires peut être estimé de manière précise en utilisant la valeur des déplacements chimiques (fréquences) sur des spectres RMN, même en l'absence d'attribution spectrale.
Le concept de structure secondaire a été introduit par Kaj Ulrik Linderstrøm-Lang (en), lors des conférences médicales Lane en 1952 à Stanford.
Protéines
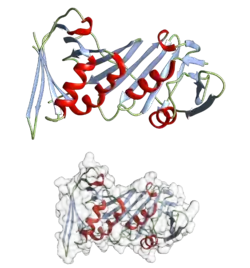
La structure secondaire des protéines consiste en un réseau d'interactions locales entre résidus d'acides aminés, par l'intermédiaire ou non de liaisons hydrogène. Les structures secondaires les plus courantes sont les hélices α et les feuillets β. D'autres types d'hélices, comme les hélices 310 et les hélices π sont prédites comme ayant des arrangements favorables de liaisons hydrogène, mais ne sont observées que de manière exceptionnelle dans les protéines naturelles, sauf à l'extrémité des hélices α. Ceci résulte d'un empilement défavorable du squelette peptidique au centre de ces hélices. D'autres structures étendues comme l'hélice polyproline et le feuillet α sont rares dans la conformation native des protéines, mais pourraient jouer un rôle important dans le processus de repliement des protéines. Des coudes, région où le squelette change brutalement de direction, et des régions flexibles et irrégulières connectent entre eux les éléments réguliers que sont les hélices et les feuillets. La pelote aléatoire n'est pas à proprement parler une structure secondaire, mais plutôt une catégorie par défaut, dans laquelle on classe les conformations qui ne correspondent pas à une structure secondaire régulière.
Les acides aminés diffèrent dans leur capacité à former les différents types de structure secondaire. La proline et la glycine sont parfois considérées comme des « briseuses d'hélice », parce qu'elles détruisent la régularité du squelette de l'hélice α. En revanche, elles ont des capacités conformationnelles particulières et se retrouvent fréquemment dans les coudes. Les acides aminés qui favorisent la formation des hélices sont la méthionine, l'alanine, la leucine, le glutamate et la lysine (« MALEK » en code acide aminé à une lettre). À l'inverse, les « gros » acides aminés aromatiques (tryptophane, tyrosine et phénylalanine) et les acides aminés branchés en Cβ (isoleucine, valine et threonine) privilégient la conformation en feuillet β. Cependant, ces tendances ne sont pas suffisamment marquées pour pouvoir servir de base à la prédiction de structure secondaire, sur la base de la seule séquence en acides aminés.
L'algorithme DSSP
Il existe plusieurs méthodes pour définir la structure secondaire d'une protéine (par exemple STRIDE[1], DEFINE[2]), mais la méthode du dictionnaire de structure secondaire de protéines (DSSP)[3] est couramment utilisée pour décrire la structure des protéines en utilisant des codes à une lettre. La structure secondaire est déterminée sur la base de l'arrangement des liaisons hydrogène, selon le schéma initial proposé par Corey et Pauling en 1951[4] (avant même qu'aucune structure de protéine ne soit résolue expérimentalement). Il existe huit types de structures secondaires définies par DSS :
- G = hélice 310Le carbonyle -CO du résidu i forme une liaison hydrogène avec l'amide du résidu i+3 (longueur minimale, 3 résidus) ;
- H = hélice α. Le carbonyle -CO du résidu i forme une liaison hydrogène avec l'amide du résidu i+4 (longueur minimale, 4 résidus) ;
- I = hélice π. Le carbonyle -CO du résidu i forme une liaison hydrogène avec l'amide du résidu i+5 (longueur minimale, 5 résidus) ;
- T = coude fermé par une liaison hydrogène (3, 4 ou 5 résidus) ;
- E = brin β étendu au sein d'un feuillet parallèle ou antiparallèle (longueur minimale, 2 résidus) ;
- B = résidu isolé dans un pont β (paire isolée formant une liaison hydrogène de type feuillet β) ;
- S = coude (sans liaison hydrogène).
Les résidus d'acides aminés qui ne sont dans aucune des conformations ci-dessus sont classés dans la huitième catégorie, « pelote », souvent représentée par ' ' (espace), C (en:Coil) ou '-' (tiret). Les hélices (G, H et I) et les feuillets doivent tous avoir une longueur minimale raisonnable. Ceci signifie que deux résidus consécutifs dans la structure doivent former le même type de liaisons hydrogène. Si l'hélice ou le feuillet est trop court, les résidus correspondant sont classés T ou B, respectivement. Il existe d'autres catégories de structures secondaires (coudes aigus, boucles oméga...), mais elles sont utilisées moins fréquemment.
Définition des liaisons hydrogène par DSSP
La structure secondaire est définie par l'arrangement des liaisons hydrogène, en conséquence, la définition exacte de celles-ci est cruciale. Dans DSSP, la définition standard d'une liaison hydrogène dérive d'un modèle purement électrostatique. DSSP attribue des charges partielles q1 de +0,42e et - 0,42e sur le carbone et l'oxygène du carbonyle (C=O) et q2 de +0,20e et - 0,20e sur l'hydrogène et l'azote de l'amide (NH), respectivement. L'énergie électrostatique est définie par :
- .
![{\displaystyle E=q_{1}q_{2}\left[{\frac {1}{r_{ON}}}+{\frac {1}{r_{CH}}}-{\frac {1}{r_{OH}}}-{\frac {1}{r_{CN}}}\right]\cdot 332\ \mathrm {kcal/mol} }](https://img.franco.wiki/i/c799ebb61003efb86023f9bb8c724673a9315d27.svg)
Selon DSSP, une liaison hydrogène existe si et seulement si E est inférieur à - 0,5 kcal/mol. Bien que le calcul utilisé par DSSP soit une approximation relativement grossière de l'énergie physique, elle est généralement acceptée pour la détermination de la structure secondaire des protéines.
Prédiction de structure secondaire des protéines
La prédiction de la structure tertiaire d'une protéine à partir de sa seule séquence en acides aminés est un problème très difficile. En revanche, la simplification permise par les définitions de structures secondaires plus restreintes ci-dessus a permis de rendre la question plus accessible et la prédiction de structure secondaire des protéines a été l'objet de recherches actives depuis de nombreuses années.
Bien que le codage DSSP à huit états soit déjà une simplification par rapport aux 20 acides aminés présents dans les protéines, la majorité des méthodes de prédiction de structure secondaire réduisent encore le problème aux trois états principaux : Hélice, Feuillet et Pelote. La manière de passer de 8 à 3 états varie suivant les méthodes. Les premières approches prédictives étaient fondées sur les propensions individuelles de chacun des acides aminés à former des hélices ou des feuillets[5], parfois couplées avec des règles pour estimer l'enthalpie libre associée à la formation de ces structures secondaires. De telles méthodes avaient une précision de l'ordre de ~60 % pour la prédiction de l'état (hélice/feuillet/pelote) adopté par un résidu. Un gain substantiel de précision (jusqu'à environ ~80 %) a été permis grâce à l'exploitation des alignements de séquences multiples entre protéines homologues. La connaissance de la distribution complète des acides aminés observée à une position donnée (et dans son voisinage, typiquement jusqu'à 7 résidus de chaque côté) au travers de l'évolution donne une image bien plus précise des tendances structurales autour de cette position. Par exemple, une protéine donnée pourrait avoir une glycine à une position donnée, ce qui, isolément, pourrait suggérer la présence d'une région en pelote. Cependant, un alignement multiple de séquences pourrait révéler que des résidus favorables à la formation d'une hélice sont présents à cette position (et aux positions voisines) dans 95 % des protéines homologues, chez des espèces distantes de près d'un milliard d'années dans l'évolution. De surcroît, en examinant l'hydrophobicité à cette position et aux positions voisines, le même alignement multiple pourrait également suggérer une distribution de l'accessibilité au solvant cohérente avec une hélice α présentant une face hydrophobe et une face hydrophile. Globalement, ces facteurs suggéreraient que le résidu glycine de la protéine étudiée fait partie d'une hélice α, plutôt que d'une région en pelote. Différents types de méthodes sont utilisées pour combiner l'ensemble des données disponibles pour formuler cette prédiction à 3 états : les réseaux de neurones, les modèles de Markov cachés ou les machines à vecteurs de support. Toutes les méthodes modernes fournissent en plus une évaluation de la fiabilité de la prédiction à chaque position (score de confiance).
Les méthodes de prédiction de structure secondaire font l'objet d'évaluations constantes, par exemple l'expérience EVA[6]. Après environ 270 semaines de test, les méthodes les plus précises sont pour l'instant PsiPRED[7], PORTER[8], PROF[9] et SABLE[10]. Le principal secteur où des gains de précision sont possibles semble être la prédiction des feuillets β. Les résidus prédits avec une confiance élevée en conformation β sont le plus souvent de manière correcte, mais les différentes méthodes disponibles ont tendance à rater certaines zones en feuillet (faux négatifs). Il est probable que la limite supérieure de la précision de ces prédictions se situe autour de ~90 %, en raison des particularités spécifiques de DSSP pour catégoriser les différentes classes de structures secondaires.
La précision de la prédiction de structure secondaire est un élément clé de la prédiction de structure tertiaire, dans tous les cas où la modélisation à partir de la structure d'une protéine homologue. Par exemple, une prédiction fiable d'un motif de six éléments de structure secondaire avec un enchaînement βαββαβ est la signature caractéristique d'un repliement de type ferrédoxine.
Acides nucléiques
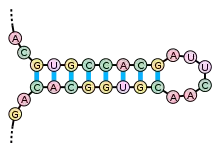
Les acides nucléiques ont aussi une structure secondaire, principalement les ARN simple-brin. La structure secondaire des ARN est divisée en hélices (paires de bases contiguës) et divers types de boucles (nucléotides non-appariés, entourés d'hélices). La structure en « tige et boucle » dans laquelle une hélice appariée se termine par une courte boucle de nucléotides non-appariés est particulièrement fréquente et constitue une brique élémentaire pour former des motifs structuraux plus complexes, comme la structure en feuille de trèfle des ARNt, où quatre hélices sont associées au niveau d'un jonction centrale. Les boucles internes (une série de nucléotides non appariées au milieu d'une hélice plus longue) et les hernies (régions où l'un des deux brins de l'hélice présente des nucléotides supplémentaires non-appariés avec le brin complémentaire) sont également très courantes. Enfin on peut observer des structures de type pseudonœud et des triplets de bases dans certains ARN.
Dans la mesure où la structure secondaire des ARN repose principalement sur la formation d'appariements de bases, celle-ci peut se définir en identifiant quels nucléotides sont appariés avec lesquels dans une molécule ou dans un complexe. Cependant les appariements canoniques de type Watson-Crick ne sont pas les seuls rencontrés, même si ce sont de loin les plus fréquents. On peut trouver des appariements bancals G-U et d'autres types de paires non canoniques (appariement Hoogsteen, en cisaille...).
Prédiction de structure d'ARN
Pour beaucoup de molécules d'ARN, la structure secondaire est essentielle à leur fonction biologique, souvent plus que leur séquence en nucléotides. Ce point constitue un atout important pour l'analyse des ARN non codants, parfois appelés « gènes ARN ». La structure secondaire des ARN peut être prédite avec certaine fiabilité par ordinateur et de nombreuses applications bio-informatiques reposent sur l'utilisation de ces techniques pour l'analyse des ARN.
La méthode générale pour la prédiction de structures secondaires d'ARN repose sur la programmation dynamique[11]. Ces méthodes sont fondées sur des calculs des enthalpies libres associées à la formation des diverses paires de bases, tabulées à partir des paramètres de stabilité thermodynamique de petits fragments d'ARN[12], mesurée grâce à l'effet hyperchrome. Cette approche ne permet cependant pas de détecter les structures non canoniques et les pseudonœuds.
Alignement
Les alignements multiples de séquences d'ARN et de protéines peuvent s'appuyer sur la connaissance de leurs structures secondaires. L'inclusion de cette information supplémentaire, quand elle est connue, permet d'améliorer sensiblement la qualité des alignements de séquences.
Dans les protéines, les insertions et délétions dans l'alignement sont en général observées au niveau des boucles et des régions en pelote, mais pas dans les hélices et les feuillets.
Dans les ARN, les appariements de bases sont en général plus conservés que la nature des bases elles-mêmes. Ainsi par exemple, on pourra trouver une paire A-U remplacée par une paire G-C dans un autre ARN homologue. La structure secondaire, sinon la séquence, est ainsi conservée et son identification est essentielle à la construction d'un alignement correct.
Notes et références
- (en) Frishman D., Argos P., « Knowledge-based protein secondary structure assignment », Proteins, vol. 23, no 4, , p. 566-579 (PMID 8749853)
- (en) Richards F. M., Kundrot C. E., « Identification of structural motifs from protein coordinate data: secondary structure and first-level supersecondary structure », Proteins, vol. 3, no 2, , p. 71-84 (PMID 3399495)
- (en) Kabsch W., Sander C., « Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features », Biopolymers, vol. 22, no 12, , p. 2577–2637 (PMID 6667333, DOI 10.1002/bip.360221211)
- (en) Pauling L., Corey R.B., Branson H.R., « Two hydrogen-bonded helical configurations of the polypeptide chain », Proc. Natl. Acad. Sci. USA, vol. 37, , p. 205-211 (PMID 14816373)
(en) Pauling L., Corey R.B., « Configurations of polypeptide chains with favored orientations of the polypeptide around single bonds: Two pleated sheets », Proc. Natl. Acad. Sci. USA, vol. 37, , p. 729-740 (PMID 16578412) - (en) Chou P.Y., Fasman G.D., « Empirical predictions of protein conformation », Annu. Rev. Biochem., vol. 47, , p. 251-276 (PMID 354496).
- « EVA »(Archive.org • Wikiwix • Archive.is • Google • Que faire ?) (consulté le ).
- PsiPRED.
- SAM, PORTER.
- PROF.
- SABLE.
- (en) Zuker M., « Computer prediction of RNA structure », Methods Enzymol., vol. 180, , p. 262–288 (PMID 2482418).
- (en) Freier S.M., Kierzek R., Jaeger J.A., Sugimoto N., Caruthers M.H., Neilson T., Turner D.H., « Improved free-energy parameters for predictions of RNA duplex stability », Proc. Natl. Acad. Sci. USA, vol. 83, , p. 9373–9377 (PMID 2432595).
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Secondary structure » (voir la liste des auteurs).
Voir aussi
Liens externes
- C. Branden et J. Tooze (1996), Introduction à la structure des protéines, De Boeck, Louvain, Belgique. (ISBN 978-2804121099)
Protéines
- Protein Data Bank banque de donnée de structure de protéines
- PROF
- Jpred
- DSSP
- WhatIf
- Rasmol (logiciel gratuit de visualisation de macromolécules, implémente DSSP)
Acides nucléiques
- « Mfold »(Archive.org • Wikiwix • Archive.is • Google • Que faire ?) (consulté le ) Prédictions de structure secondaire d'acides nucléiques