Protéine tau
La protéine tau (en anglais : tubulin-associated unit) est une protéine animale. Elle fait partie de la famille des protéines associées aux microtubules (protéines MAP). Chez les humains, ces protéines sont surtout présentes dans les neurones par rapport aux cellules non neuronales du système nerveux central. Une des principales fonctions des protéines tau est d'interagir avec la tubuline afin de moduler la stabilité des microtubules des axones.
Tau peut être présente dans les dendrites et est principalement active dans les portions distales des axones où elle permet une stabilisation des microtubules, mais également leur flexibilité nécessaire. Cela contraste avec les protéines MAP6 (STOP) dans les portions proximales d'axones qui ont pour fonction essentielle de verrouiller les microtubules et avec les protéines MAP2 qui stabilisent les microtubules dans les dendrites.
| Protéine tau | ||
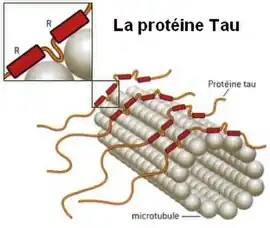 Schéma de la protéine tau assurant la cohésion des microtubules du cytosquelette des neurones. | ||
| Caractéristiques générales | ||
|---|---|---|
| Nom approuvé | Microtubule Associated Protein Tau | |
| Symbole | MAPT | |
| Classification | ||
| Pfam | PF00418 | |
| InterPro | IPR001084 | |
| PROSITE | PDOC00201 | |
| Homo sapiens | ||
| Locus | 17q21.31 | |
| Masse moléculaire | 78 928 Da[1] | |
| Nombre de résidus | 758 acides aminés[1] | |
| Liens accessibles depuis GeneCards et HUGO. | ||
Découverte
Les protéines Tau ont été découvertes comme des protéines favorisant la polymérisation des dimères de tubuline en microtubules en 1975 à la suite des travaux de Marc Kirschner[2]. En 1963, en étudiant des neurones en dégénérescence dans la maladie d'Alzheimer, des chercheurs découvrent des accumulations de structures filamenteuses dans leur cytoplasme. Ces structures, dénommées PHF (Paired Helical Filaments), sont en fait constituées de protéines tau comme l'indique, en 1985, le travail de l'équipe de Jean-Pierre Brion[3].
Les maladies neurologiques dont les symptômes sont imputables à des dysfonctionnements de tau, sont qualifiées de taupathies (ou tauopathies). La plus connue est la maladie d'Alzheimer.
Tau semble être impliquée dans la signalisation à l’insuline au niveau du cerveau, indispensable à la mémoire et à l’homéostasie glucidique[4] - [5]. Tau est aussi impliqué dans la protection des acides nucléiques (ADN et ARN) dans des conditions de stress cellulaire. Ces conclusions de recherches[6] - [7] sur cette protéine, ouvrent la voie à de nouvelles pistes thérapeutiques.
Fonction et codage
Les protéines sont codées par les gènes situés sur nos chromosomes, et leur codage obéit à des mécanismes biochimiques et moléculaires complexes, à dessein d'assumer des fonctions normales de vie ou de mort (apoptose) de nos cellules. Les protéines tau sont le produit d'épissage alternatif à partir d'un gène unique. Ce gène codant la protéine tau est situé sur le bras long du chromosome 17 à la position 17q21, il est formé de 16 exons qui s'étendent sur 100 kpb.
La fonction des protéines tau est d’interagir avec les microtubules via des domaines spécifiques de liaison et de favoriser l'assemblage et la stabilité des microtubules. L'interaction de la protéine tau avec les microtubules est régulée par phosphorylation. Tau est une phosphoprotéine qui contient environ 85 sites potentiels de phosphorylation (isoforme 2N4R du cerveau humain - 441 acides aminés).
Les protéines Tau ont deux manières de contrôler la stabilité des microtubules : la phosphorylation et les isoformes.
Tau, des phosphoprotéines
La phosphorylation de la protéine tau est régulée par un grand nombre de kinases. Par exemple, PKN, une sérine / thréonine kinase. Lorsque PKN est activée, elle phosphoryle la protéine tau, ce qui entraîne une perturbation de l'organisation des microtubules. Tau est une phosphoprotéine qui contient environ 80 sites potentiels de phosphorylation. La régulation de l’état de phosphorylation de la protéine tau résulte des activités conjointes de protéines kinases et de protéines phosphatases.
En général, une hyperphosphorylation de la protéine tau diminue son affinité pour les microtubules, ce qui peut entraîner leur déstabilisation et par conséquent une désorganisation du cytosquelette. Or, une perturbation du cytosquelette intervient au cours de l’apoptose neuronale, indiquant que des modifications de l’état de phosphorylation de la protéine tau pourraient jouer un rôle important dans la mort neuronale par apoptose.
L'hyperphosphorylation de la protéine tau (inclusions tau, pTau) peut entraîner l'auto-assemblage des enchevêtrements de paires de filaments hélicoïdaux et des filaments droits, qui sont impliqués dans la pathogenèse de la maladie d'Alzheimer et d'autres tauopathies[8].
Tau, des protéines isoformes
La protéine tau majeure dans le cerveau humain est codée par 11 exons. Les isoformes sont le résultat de l'épissage alternatif dans les exons 2, 3, et 10 du gène tau, laissant six combinaisons (2 - 3 - 10 - (0N3R); 2 + 3 - 10 - (1N3R); 2 + 3 + 10 - (2N3R); 2 - 3 - 10 + (0N4R); 2 + 3 - 10 + (1N4R); 2 + 3 + 10 + (2N4R)). Ainsi, dans le cerveau humain, les protéines tau constituent une famille de six protéines isoformes dépendantes d'un épissage alternatif, avec la gamme de 352 à 441 acides aminés.
Les 6 tau isoformes se distinguent donc par leur nombre de domaines de liaison : trois isoformes ont trois domaines de liaison (3R) et les trois autres ont quatre domaines de liaison (4R). Les domaines de liaison ont des situations différentes et sont chargés positivement (ce qui permet de se lier aux microtubules de charge négative). Les isoformes à quatre domaines de liaison stabilisent mieux les microtubules que celles avec trois domaines de liaison.
Les tau isoformes diffèrent dans les inserts de 0, 1 ou 2, soit de 29 acides aminés à la partie N-terminale (exons 2 et 3), et 3 ou 4 régions répétées à l'extrémité C-terminale dans l'exon 10 disparus. Ainsi, l’isoforme la plus longue dans le SNC a quatre répétitions (R1, R2, R3 et R4) et deux inserts (441 acides aminés au total), tandis que l’isoforme la plus courte a trois répétitions (R1, R3 et R4) et aucun insert (352 acides aminés au total). Si on considère l'isoforme la plus longue du cerveau humain, elle contient 85 sites potentiels de phosphorylation.
Le gène MAPT a deux haplogroupes, H1 et H2, dans lesquels le gène apparaît dans des orientations inversées. L’haplogroupe H2 est commun seulement en Europe et chez les personnes d'ascendance européenne. L’haplogroupe H1 semble être associé avec une probabilité accrue de certaines démences, comme la maladie d'Alzheimer.
Interactions
Il a été démontré que la protéine tau interagissait avec les protéines FYN (en)[9], alpha-synucléine[10] - [11], YWHAZ (en)[12] et S100B (en)[13] - [14].
| Synucléines | Protéines tau | |
|---|---|---|
| Localisation | Petites protéines spécialement abondantes dans les neurones et surtout dans les terminaisons pré-synaptiques. | Les protéines tau (Tubule Associated Unit) appartiennent au groupe des MAP (Microtubule Associated Proteins). |
| Type | Il existe 3 types de synucléines : alpha, béta et gamma. Le gène de l'alpha-synucléine, PARK1 est situé sur le bras long du chromosome 4. | Il existe 6 isoformes de protéines tau dans le cerveau adulte humain, dépendant d'un épissage alternatif de 3 exons de l'ARN d'un même gène situé sur le bras long du chromosome 17. |
| Rôle | Encore imprécis à ce jour. | Le rôle essentiel des protéines associées aux microtubules est la stabilisation de leurs composants du cytosquelette. Les microtubules servent à transporter les matériaux synthétisés par le corps cellulaire vers les terminaisons nerveuses et à en maintenir la forme. |
| Processus pathologique | Réduction de la solubilité des alpha-synucléines. | Modifications post-traductionnelles comme la phosphorylation ou la troncation de différentes combinaisons d’isoformes. |
| Conséquences | Transformation en agrégats ou en filaments d’allure amyloïde. | Transformation en différents types de filaments selon les combinaisons d’isoformes. |
| Pathologies associées | Parkinson, Démences avec corps de Lewy et Atrophie Multi-Systématisée (AMS). | Alzheimer, PSP (paralysie supranucléaire progressive), DCB (dégénérescence corticobasale), maladie des grains argyrophiles (AGD)[15], maladie de Pick et DFT (dégénérescences frontotemporales, avec syndrome parkinsonien, liées au chromosome 17 DLFT17). |
Tauopathies ou taupathies

L'agrégation des protéines tau est rencontrée dans de nombreuses maladies neurodégénératives et autres pathologies (neuromusculaire, métabolique...), appelées tauopathies[16]. Si les six isoformes de protéines s'agrègent dans la maladie d'Alzheimer, ce n'est pas le cas dans d'autres tauopathies comme la PSP/DCB où les isoformes à quatre domaines de liaison aux microtubules s'agrègent de façon préférentielle. À l'inverse, dans la maladie de Pick, une dégénérescence lobaire frontotemporale, seules des isoformes à trois domaines de liaison aux microtubules sont retrouvées agrégées au sein des corps de Pick[17]. De très nombreuses maladies sont liées à des dérèglements de mécanismes contrôlant l’apoptose. Toute anomalie de l’apoptose peut déclencher ou accélérer de nombreuses pathologies caractérisées par un déficit ou à l’inverse par une activation inappropriée des mécanismes apoptotiques. Pourtant l’apoptose est une mort cellulaire programmée qui ne laisse habituellement pas d'agrégats. Or dans les Tauopathies, la mort neuronale conduit à une agrégation de protéines Tau, signant une apoptose anormale ou pathologique[18].
La dégénérescence neurofibrillaire rencontrée dans la maladie d'Alzheimer suit un processus très hiérarchisé. Elle débute dans la formation hippocampique, puis le lobe temporal et les autres régions polymodales associatives, ensuite les régions unimodales et enfin l'ensemble du cortex cérébral. Les régions primaires sont les dernières touchées. Cette séquence de neurodégénérescence pourrait s'expliquer par une vulnérabilité sélective de sous-populations neuronales et/ou une propagation de type prion[19].
En 1993, ApoE4 (variant muté du gène de l'apolipoprotéine E) s'est montrée capable de multiplier par 4 le risque de survenue de la forme la plus commune de la maladie d'Alzheimer ; c'est le risque génétique le plus prédictif pour la maladie. On a donc recherché d'éventuels liens entre ApoE4 et la protéine β-amyloïde (1er suspect comme cause de la maladie), mais plutôt vainement jusqu'en 2017. Là David Holtzman, l'un des principaux défenseurs de l'hypothèse d'une ApoE4 exacerbant la pathologie amyloïde a surpris la communauté scientifique : avec une large équipe de chercheurs il montre[20] que les effets les plus toxiques de l'ApoE4 résulteraient non pas de son action sur l'amyloïde, mais d'une réponse immunitaire dommageable à une autre protéine : Tau (cette protéine normalement stabilisante dans les cellules semble pouvoir échapper aux neurones ; elle peut prendre des formes pathologiques pouvant se propager d'une cellule à l'autre ; le système immunitaire lance alors une réponse inflammatoire qui en laboratoire et in vitro s'avère capable de massivement tuer des neurones).
Les stratégies thérapeutiques hésitaient depuis longtemps entre viser tau ou l'amyloïde. Cette étude montre que ces deux protéines sont ciblées par le gène muté ApoE4, et que la β-amyloïde joue un premier rôle dans le déclenchement de la maladie d'Alzheimer, et que les dépôts de tau créent ensuite de nouveaux dégâts... L'ApoE4 pourrait donc être une clé pour comprendre et traiter cette maladie (et d'autres « tauopathies » pouvant aussi découler d'anomalies de l'ApoE induisant une réponse immunitaire innée capable de tuer les neurones au lieu de les défendre). Les partisans de l'hypothèse Tau et de l'hypothèse amyloïde peuvent se réconcilier et travailler à maitriser les dégâts de l'ApoE4 qui s'exercent sur ces deux protéines vitales[21].
Pathologies liées à la protéine tau

Les études postmortem de cerveaux de patients atteints d'une maladie neurodégénérative (maladie d'Alzheimer, PSP, DCB, Pick, DFT, Syndromes Parkinsoniens Atypiques, etc.) montrent la dégénérescence des cellules nerveuses du cerveau en un certain nombre de zones où la présence de protéines tau pathogènes est systématique, pouvant notamment être associée à une mutation du chromosome 17 (FTDP-17)[24] - [25] - [26] - [27]. Cette présence se caractérise par des agrégats anormaux de cette protéine, sans qu'il soit établi à ce jour, s'ils sont la cause ou la conséquence de la mort cellulaire.
Les études post-mortem démontrent aussi une forte corrélation positive entre l'augmentation de la protéine Tau dans le lobe pariétal et l'altération des fonctions cognitives ante-mortem[8] - [28].
Parmi les maladies dites de Parkinson plus (syndromes parkinsoniens atypiques), la PSP est une tauopathie, qui peut être qualifiée de « pure » dans la mesure où cette maladie semble mettre en jeu essentiellement un mécanisme pathogène lié à la protéine tau (ou à sa phosphorylation), ce qui n'est pas le cas par exemple pour la maladie d'Alzheimer, autre tauopathie pour laquelle la protéine tau est aussi très étudiée. Dans la maladie d'Alzheimer à un stade de l'évolution de la pathologie tau, il y a l'apparition de substances amyloïdes, puis de plaques amyloïdes qui signent cette maladie. Les mécanismes qui président dans la maladie d'Alzheimer à l'apparition de la première plaque amyloïde, concomitante au développement de la pathologie tau, ne sont pas connus, pas plus que les processus tau et amyloïdes de leurs développements au sein du système nerveux central.
Perspectives thérapeutiques
- Il existe de nombreuses stratégies thérapeutiques ciblant la protéine tau. Historiquement, les approches thérapeutiques ont visé à diminuer la phosphorylation de tau ou à stabiliser les microtubules. Les suivantes ont cherché à empêcher l'agrégation[29]. Les dernières visent à faciliter la dégradation de la protéine en modulant l'autophagie ou le protéasome ou par immunothérapie[30] et à diminuer son expression par des approches oligonucléotides anti-sens[31].
- le semorinemab est un anticorps monoclonal dirigé contre la protéine Tau. Il n'a cependant pas démontré d'efficacité sur l'évolutivité de la maladie d'Alzheimer[32].
- Des chercheurs[33] du Minnesota, en stimulant la production de protéines tau ont réussi à créer une démence proche de la maladie d'Alzheimer chez des souris : leurs résultats aux tests de mémoire chutaient rapidement. Quand ils ont arrêté la production de nouvelles protéines dans le cerveau des souris, leur mémoire a cessé de se détériorer et les performances aux tests ont commencé à s'améliorer. Le cerveau murin serait donc capable de récupérer une partie du terrain que les protéines tau lui avaient fait perdre. (Marion Garteiser, journaliste santé - 02/08/2005). Cette seule expérience ne permet pas de dire si elle est reproductible chez l'être humain, mais c'est un résultat encourageant, si l'on considère que jusqu'à la date de ces expériences, confirmées par d'autres depuis, il était tenu pour irréversibles les pertes de capacités intellectuelles dues aux maladies neurodégénératives comme celle d'Alzheimer ou de la PSP.
- Le , le Pr Beaulieu a annoncé avoir avec son équipe caractérisé l’interaction entre la protéine tau et une autre protéine (la FKBP52[34]) naturellement très abondante dans le cerveau. Ils ont démontré in vitro que la protéine FKBP52 supprimait l’activité de la protéine tau. En d'autres mots, une forte expression de FKBP52 empêche l’accumulation de protéines tau dans les cellules nerveuses.
Cette avancée en recherche fondamentale très prometteuse permet d'envisager :- de pouvoir mesurer la quantité de protéines FKBP52 chez les personnes pour évaluer leur risque ultérieur de développer une tauopathie (Alzheimer, PSP, DCB, Pick, DFT, Syndromes Parkinsoniens Atypiques, etc.).Une étude devrait bientôt commencer.
- de trouver des médicaments capables de stimuler cette protéine anti-tau, même si d'autres médicaments[35] « anti-tau » ou « anti-amas de protéine tau », agissant de façon différente, sont actuellement expérimentés[36] chez l’homme.
- Chez une lignée[37] de souris transgéniques ayant de graves difficultés de l'apprentissage moteur, l'administration orale de méthylthioninium (MT, composant du bleu de méthylène) compense ce déficit et réduit la charge de tau, attribué à la probable activation des fonctions d'autophagie[38].
Chez l'Homme, une étude clinique contrôlée de 321 malades d'Alzheimer a montré l'inhibition de l’agrégation de tau, accompagnée d'une réduction significative du déclin cognitif clinique (de 84 % sur 50 semaines, mesuré par l'échelle ADAS-cog[39]), qui reste à confirmer par imagerie neurologique. Le MT semble donc potentiellement utile aux stades précoces de la maladie et en 2011 un variant amélioré de MT devait être testé en études de phase 3[40]. En 2020, il n'existe toujours pas de traitement reconnu basé sur ces propriétés, mais les études se poursuivent aussi bien sur des dérivés du bleu de méthylène (Hydromethanesulphonate)[41] que via d'autres substances susceptibles de réguler la voie autophagique [42]
Notes et références
- Les valeurs de la masse et du nombre de résidus indiquées ici sont celles du précurseur protéique issu de la traduction du gène, avant modifications post-traductionnelles, et peuvent différer significativement des valeurs correspondantes pour la protéine fonctionnelle.
- Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW (mai 1975). « Un facteur protéique essentiel pour l'assemblage des microtubules. » Proc. Natl. Acad. Sci. USA.
- Jean-Pierre Brion (Bruxelles, Belgique)
- INSERM - Maladie d'Alzheimer : la protéine Tau serait aussi impliquée dans les troubles métaboliques
- Elodie Marciniak et al., Tau deletion promotes brain insulin resistance, 2017. DOI 10.1084/jem.20161731
- Tau impliquée dans la dégénérescence neuronale serait capable de protéger l'ADN
- Marie Violet, Lucie Delattre, Meryem Tardivel et Audrey Sultan, « A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions », Frontiers in Cellular Neuroscience, vol. 8, (ISSN 1662-5102, PMID 24672431, PMCID PMC3957276, DOI 10.3389/fncel.2014.00084, lire en ligne, consulté le )
- (en) Tremblay C., François A, Delay C, Freland L, Vandal M, Bennett DA et Calon F, « Association of Neuropathological Markers in the Parietal Cortex With Antemortem Cognitive Function in Persons With Mild Cognitive Impairment and Alzheimer Disease. Journal of neuropathology and experimental neurology », J Neuropathol Exp Neurol, , p. 70-88 (ISSN 1554-6578, lire en ligne)
- Klein, Corinne; Kramer Eva-Maria, Anne-Marie Cardine, Schraven Burkhardt, Brandt Roland, Jacqueline Trotter (février 2002 ). « Excroissance du processus d'oligodendrocytes est promu par l'interaction de la kinase Fyn avec la protéine du cytosquelette tau ». J. Neurosci (États-Unis)
- Jensen, PH; Hager H, Nielsen MS, Hojrup P, J Gliemann, Jakes R (septembre 1999 ). « Alpha-synucléine se lie à tau et stimule la protéine kinase A catalysée phosphorylation de tau de résidus sérine 262 et 356 ». J. Biol. Chem (ÉTATS-UNIS).
- BI Giasson, Lee VM, Trojanowski JQ (2003). « Interactions des protéines amyloïdogéniques » Neuromolecular Med.
- Hashiguchi, M; Sobue K, Paudel HK (août 2000 ). « 14-3-3zeta est un effecteur de la phosphorylation de la protéine tau ». J. Biol. Chem (ÉTATS-UNIS).
- Yu, WH; Fraser, PE (avril 2001 ). « L'interaction avec les S100beta tau est promu par le zinc et inhibée par l'hyperphosphorylation de la maladie d'Alzheimer ». J. Neurosci (États-Unis).
- Baudier, J; Cole RD (avril 1988 ). « Les interactions entre les protéines associées aux microtubules tau et S100B réguler la phosphorylation de tau par le Ca2 + / calmoduline-dépendante protéine kinase II ». J. Biol. . Chem (ÉTATS-UNIS).
- Démence à grains argyrophiles (Argyrophilic Grain Disease ou AGD)
- Lebouvier, Pasquier et Buée, « Update on tauopathies », Current Opinion in Neurology, , p. 589-598 (ISSN 1350-7540, lire en ligne)
- (en) Buée-Scherrer, Hof et Buée et al., « Hyperphosphorylated tau proteins differentiate corticobasal degeneration and Pick’s disease », Acta Neuropathologica, , p. 351-359 (ISSN 1432-0533, lire en ligne)
- Malika Hamdane, Patrice Delobel, Anne-Véronique Sambo et Caroline Smet, « Neurofibrillary degeneration of the Alzheimer-type: an alternate pathway to neuronal apoptosis? », Biochemical Pharmacology, vol. 66, no 8, , p. 1619–1625 (ISSN 0006-2952, PMID 14555242, lire en ligne, consulté le )
- « Traiter la maladie de tau », sur Cerveau&Psycho, Cerveau&Psycho, (consulté le )
- Yang Shi & al. (2017) ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy , Nature | doi:10.1038/nature24016
- Underwood Emily (2017) A new study is changing how scientists think about Alzheimer’s disease ; Science News, 20 sept 2017
- Delacourte A, David JP, Sergeant N, et al. « The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer's disease ». Neurology 1999 ; 52 : 1158-65.
- Delacourte A, Buée L. « Normal and pathological Tau proteins as factors for microtubule assembly ». Int Rev Cytol 1997 ; 171 : 167-224.
- Kowalska A., The genetics of dementias. Part 1: Molecular basis of frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) ; Postepy Hig Med Dosw (Online). 2009 Jun 15; 63:278-86. Epub 2009 Jun 15.
- Spillantini MG, Van Swieten JC, Goedert M, Tau gene mutations in frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). . Neurogenetics. 2000 Mar; 2(4):193-205 (résumé)
- Goedert M, Spillantini MG., Tau gene mutations and neurodegeneration. Biochem Soc Symp. 2001; (67):59-71.
- Goedert M, Ghetti B, Spillantini MG., Tau gene mutations in frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). Their relevance for understanding the neurogenerative process. Ann N Y Acad Sci. 2000; 920:74-83
- (en) Tremblay C, Pilote M, Phivilay A, Emond V, Bennett DA et Calon F, « Biochemical Characterization of Aβ and Tau Pathologies in Mild Cognitive Impairment and Alzheimer's Disease », Journal of Alzheimer's Disease 12, , p. 377-390 (ISSN 1387-2877, lire en ligne)
- « Traiter la maladie de « tau » », sur https://www.cerveauetpsycho.fr, (consulté le )
- (en) COLIN, « From the prion-like propagation hypothesis to therapeutic strategies of anti-tau immunotherapy », Acta Neuropathologica, , Acta Neuropathologica volume 139, pages 3–25 (2020) (lire en ligne
 [html])
[html]) - (en) « BIIB080 » [html], sur Alzforum, (consulté le )
- Teng E, Manser PT, Pickthorn K et al. Safety and Efficacy of Semorinemab in Individuals With Prodromal to Mild Alzheimer Disease: A Randomized Clinical Trial, JAMA Neuro, 2022;doi:10.1001
- étude de l'Université du Minnesota, menée sur des souris
- [PDF] cpINSERM 26-janv-2010
- [PDF] Zeltia 16-sept-2010
- Stratégie ciblant la protéine Tau, Prise en charge de la maladie d'Alzheimer : données actuelles et en cours de développement - Mars 2011 Extraits
- « Lignée 66 » ; souris transgénique à phénotype moteur de type frontotemporal avec abondance de tau filamentaire
- Erin E. Congdon, Jessica W. Wu, Natura Myeku et Yvette H. Figueroa, « Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo », Autophagy, vol. 8, no 4, , p. 609–622 (ISSN 1554-8627, PMID 22361619, PMCID 3405840, DOI 10.4161/auto.19048, lire en ligne, consulté le )
- Les échelles en gériatrie, PDF du CHU de Rouen
- L'écho des souris, voir chapitre «Les effets du méthylthioninium chez les souris tau transgéniques ont eu un rôle décisif dans l’encouragement de la recherche clinique», consulté 2011-11-06
- Gordon K. Wilcock, Serge Gauthier, Giovanni B. Frisoni et Jianping Jia, « Potential of Low Dose Leuco-Methylthioninium Bis(Hydromethanesulphonate) (LMTM) Monotherapy for Treatment of Mild Alzheimer's Disease: Cohort Analysis as Modified Primary Outcome in a Phase III Clinical Trial », Journal of Alzheimer's disease: JAD, vol. 61, no 1, , p. 435–457 (ISSN 1875-8908, PMID 29154277, PMCID 5734125, DOI 10.3233/JAD-170560, lire en ligne, consulté le )
- M. Catarina Silva, Ghata A. Nandi, Sharon Tentarelli et Ian K. Gurrell, « Prolonged tau clearance and stress vulnerability rescue by pharmacological activation of autophagy in tauopathy neurons », Nature Communications, vol. 11, (ISSN 2041-1723, PMID 32591533, PMCID 7320012, DOI 10.1038/s41467-020-16984-1, lire en ligne, consulté le )
Voir aussi
Articles connexes
Bibliographie
- Deming, Y. et al. (2017) Genome-wide association study identifies four novel loci associated with Alzheimer’s endophenotypes and disease modifiers. Acta Neuropathol. 133, 839–856
- Janke C, Beck M, Stahl T, Holzer M, Brauer K, Bigl V, Arendt T. (1999), Phylogenetic diversity of the expression of the microtubule-associated protein tau: implications for neurodegenerative disorders ; Brain Res Mol Brain Res. 7;68(1-2):119-28 (résumé)
- Mishra, A. et al. (2017) Gene-based association studies report genetic links for clinical subtypes of frontotemporal dementia. Brain 140, 1437–1446
- Simic G et coll. (2016) Tau protein hyperphosphorylation and aggregation in Alzheimer's disease and other tauopathies, and possible neuroprotective strategies. Biomolecules, 6(1), 6; doi:10.3390/biom6010006