Létalité synthétique
La létalité synthétique est un cas de mort cellulaire résultant de la déficience de deux ou plusieurs gènes, aucune déficience d'un seul de ces gènes n'ayant cet effet, que ces déficiences proviennent elles-mêmes de mutations, de modifications épigénétiques ou de l'action inhibitrice d'un des gènes concernés.
Lors d'un dépistage génétique de létalité synthétique, il est nécessaire de partir d'une mutation qui ne tue pas la cellule, qui peut éventuellement conférer un certain phénotype (par exemple une croissance ralentie), et ensuite tester de façon systématique d'autres mutations à d'autres loci afin de déterminer lequel confère cette létalité. La létalité synthétique est utile en thérapie cancéreuse ciblant les molécules, avec l'exemple de la thérapie ciblant les molécules exploitant un gène létal synthétique exposé par un gène suppresseur de tumeurs inactivé (BRCA1 et 2) qui a reçu l'aval de la FDA en 2016 (inhibiteur de PARP). La létalité collatérale constitue un sous-cas de létalité synthétique, où les vulnérabilités sont exposées par la délétion de gènes passagers plutôt que par un suppresseur de tumeurs[1].
Contexte
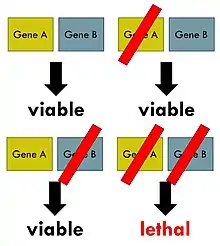
Le phénomène de létalité synthétique a été décrit pour la première fois par Calvin Bridges en 1992, qui a remarqué que certaines combinaisons de mutations chez l'organisme modèle Drosophila melanogaster conféraient une létalité[2]. Theodosius Dobjansky a formulé le terme de synthetic lethality en 1946 afin de décrire le même type d'interaction génétique chez des souches sauvages de Drosophila[3]. Si la combinaison d'événements génétiques résulte en une diminution non létale de la fitness, l'interaction est appelée "maladie synthétique". Bien qu'en génétique classique le terme de létalité fait référence à l'interaction entre deux perturbations génétiques, la létalité synthétique peut aussi s'appliquer aux cas où la combinaison d'une mutation et de l'action d'un composé chimique cause une létalité, alors que la mutation ou le composé pris séparément n'est pas létal(e)[4].
La létalité synthétique est la conséquence d'une tendance des organismes à maintenir des "schémas de mise en mémoire tampon" permettant une stabilité phénotypique malgré des variations génétiques, des changements environnementaux et des événements aléatoires tels que les mutations. Cette robustesse génétique est le résultat de voies redondantes parallèles et de "protéines condensatrices" qui camouflent les effets des mutations de telle façon que les processus cellulaires importants ne dépendent d'aucun composant individuel[5]. La létalité synthétique peut aider à identifier ces liens de "mise en mémoire tampon", ainsi que le type de maladie ou dysfonctionnement quand ces liens se décomposent, grâce à l'identification d'interactions géniques qui fonctionnent soit dans le même processus biochimique soit dans des voies métaboliques qui se révèlent ne pas être liées[6].
Tests à haut débit
La létalité synthétique peut être explorée dans une large gamme d'organismes modèles, dont Drosophila melanogaster et Saccharomyces cerevisiae. Comme les mutations létales synthétiques ne sont pas intrinsèquement viables, les approches communes consistent à utiliser des mutations thermosensibles ou insérer des mutations sous le contrôle d'un promoteur régulé afin de permettre l'exploration du phénotype résultant sans conduire à la mort de l'organisme[7]. Certaines paires de gènes létaux synthétiques peuvent être détectées en essayant d'élucider des mécanismes moléculaires de processus biologiques fondamentaux sans utiliser de tests à haut débit[8]. Par exemple, la létalité synthétique de la parkine et de la MTF1 chez Drosophila a été découverte en examinant le lien entre un stress oxydatif et l'homéostasie métallique dans la pathogénie de la maladie de Parkinson.
Cependant, des tests à haut débit de létalité synthétique peuvent aider à mieux comprendre la façon dont les processus cellulaires fonctionnent sans connaissance préalable de la fonction du gène ou de l'interaction. La stratégie de dépistage doit prendre en compte l'organisme utilisé, le mode de perturbation génétique, et si le dépistage est classique ou inverse. La plupart des premiers tests de létalité synthétique ont été réalisés chez S. cerevisiae ; en effet, la levure bourgeonnante possède plusieurs avantages dans les tests, comme le fait d'avoir un petit génome, un temps de doublement court, un état haploïde et un état diploïde, et une facilité de manipulation génétique[9]. L'ablation d'un gène peut être réalisée en utilisant une stratégie basée sur des PCR et des banques complètes de knock-out pour les gènes annotés de levure qui sont libres d'accès. La matrice génétique synthétique (synthetic genetic array ou SGA en anglais), la létalité synthétique sur microréseau (synthetic lethality by microarray ou SLAM en anglais) et la cartographie d'interaction génétique (genetic interaction mapping ou GIM en anglais) constituent trois méthodes à haut débit permettant d'analyser la létalité synthétique chez la levure. Une carte d'interaction génétique à l'échelle du génome a été créée à la suite d'une analyse SGA chez S. cerevisiae qui comprend environ 75 % de tous les gènes de la levure[10]. En examinant la létalité synthétique de 5,4 millions de paires de gènes, un réseau de connexions fonctionnelles entre les interactions génétiques a été construit.
Les dépistages à haut débit de létalité synthétique sont aussi réalisés chez les métazoaires, mais le problème principal rencontré est l'efficacité de la perturbation génique. Chez le nématode C. elegans, l'interférence par ARN peut être combinée avec une mutation de perte de fonction chez une souche d'intérêt. Alors que l'interférence par ARN est plus contraignante expérimentalement chez Drosophila, les microréseaux de cellules vivantes permettent d'inactiver deux gènes simultanément[11]. L'interférence par ARN est aussi applicable aux cellules de mammifères, et les tests génétiques dans des lignées de cellules de mammifères sont importants pour identifier des cibles pharmacologiques de médicaments.
Chimiothérapies
Une approche en termes de létalité synthétique est actuellement explorée en thérapie cancéreuse comme moyen de développer des traitements qui réduisent les effets secondaires des chimiothérapies et des médicaments chimiopréventifs. Les cellules cancéreuses sont marquées par une instabilité génétique, des erreurs lors de la réparation de l'ADN, et une transcription non contrôlée, ce qui crée des nouveaux partenaires (les deux formant des paires) létaux synthétiques dans les cellules cancéreuses. Parce que l'effet d'un médicament ciblant un produit génique spécifique ressemble au phénotype causé par une mutation dans ce même gène, une mutation liée à un cancer peut sensibiliser des cellules cancéreuses à des chimiothérapies qui ciblent son partenaire létal synthétique. Par conséquent, des médicaments qui ciblent des partenaires létaux synthétiques dans les cellules cancéreuses pourraient ne pas être cytotoxiques pour des cellules saines, ce qui permettrait d'éviter les effets secondaires des chimiothérapies.
L'analyse de la létalité synthétique peut être utilisée afin d'élucider des mécanismes de médicaments thérapeutiques connus en identifiant des gènes dont la fonction est nécessaire pour la fonction du médicament. Par exemple, BRCA1 et BRCA2 sont importants pour réparer des cassures double brin dans l'ADN, et des mutations au sein de ces gènes prédisposent les individus qui les portent au cancer du sein et au cancer des ovaires[12]. L'enzyme PARP1 est impliquée dans la réparation des cassures simple brin, et l'inhibition de la PARP1 chez un BRCA muté est sélectivement létale pour les tumeurs parce que les cellules cancéreuses accumulent des lésions dans l'ADN qu'elles ne peuvent pas réparer. La létalité synthétique est aussi utile pour tester des banques de molécules afin de détecter des médicaments qui inhibent de façon sélective les cellules cancéreuses. Lors d'un dépistage chimio-génétique effectué récemment, l'un des composés des 3 200 molécules testées était un inhibiteur létal synthétique des cellules de cancer du pancréas "gain de fonction" exprimant la KRAS, ce qui laisse entrevoir un traitement potentiel pour ce type de cancer[13].
Source des mutations et épimutations cancérigènes
Comme soulevé par Gao et ses collaborateurs[14], la stabilité et l'intégrité du génome humain sont maintenues par le système de réparation des dommages à l'ADN (DNA damage repair ou DDR en anglais). La présence d'un défaut dans le système DDR entraîne une accumulation des dommages dans l'ADN. Un dommage dans l'ADN non réparé constitue la cause majeure des mutations qui conduisent à des cancers[15] - [16]. Un tel excès de dommages dans l'ADN peut augmenter les mutations lors de la réplication de l'ADN à cause d'une tendance à faire des erreurs lors de la synthèse de translésion (voir section 2.4 de l'article sur la réparation de l'ADN en anglais). Un excès de dommages dans l'ADN peut aussi augmenter les modifications épigénétiques en raison d'erreurs lors de la réparation de l'ADN[17] - [18]. De telles mutations ainsi que des modifications épigénétiques[19] causent la progression vers un cancer.
Importance des déficiences du système DDR dans les cancers
Environ 3 ou 4 mutations "pilotes" et 60 mutations "passagères" se produisent dans l'exome (région codant des protéines) lors d'un cancer[19]. Cependant, un nombre beaucoup plus important de mutations se produisent dans les régions non codantes lors d'un cancer. Le nombre moyen de mutations dans la séquence d'ADN dans l'entièreté du génome d'un échantillon de tissu de cancer du sein est beaucoup plus important, soit environ 20 000[20]. Dans un tissu moyen de mélanome, le nombre total de mutations dans l'ADN est environ de 80 000[21].
Des modifications épigénétiques ou des mutations dans les gènes du système DDR ont de fortes chances d'être la source d'une instabilité génétique caractéristique des cancers[22]. Alors qu'une mutation ou un événement épigénétique dans un gène DDR, en tant que tel, ne conférerait pas d'avantage sélectif, un tel défaut de réparation pourrait être emporté comme une mutation "passagère" dans une cellule lorsque celle-ci acquiert une mutation/épimutation supplémentaire qui apporte un avantage pour la prolifération. De telles cellules, avec à la fois des avantages prolifératifs et un ou plusieurs défauts de réparation de l'ADN (qui causent un taux de mutation très important), ont tendance à donner naissance aux 20 000 à 80 000 mutations génomiques fréquemment observées dans les cancers. Ainsi, un défaut dans un gène DDR aura beaucoup de chances d'être présent dans les cancers (voir par exemple les fréquences des épimutations dans les gènes de réparation de l'ADN dans les cancers). La létalité synthétique avec un défaut identifié dans la réparation de l'ADN dans un cancer pourrait ainsi constituer une méthode fréquente efficace dans l'attaque thérapeutique du cancer.
Létalité collatérale
La létalité collatérale est un sous-cas de létalité synthétique dans la thérapie personnalisée contre le cancer, dans laquelle les vulnérabilités sont exposées par la délétion de gènes "passagers" plutôt que des gènes suppresseurs de tumeurs, qui sont supprimés grâce à la proximité chromosomique des principaux loci suppresseurs de tumeurs qui ont été supprimés[1].
ENO1/ENO2
La délétion homozygote du locus 1p36, qui contient plusieurs gènes candidats comme suppresseurs de tumeurs, se produit dans environ 5 % des glioblastomes, des carcinomes hépatocellulaires et des cholangiocarcinomes. La délétion de cette région inclut souvent la délétion de plusieurs gènes « passagers », dont ENO1, qui a été identifié comme conférant une létalité collatérale suivie d'une inhibition de son paralogue redondant ENO2. ENO1 encode l'isoforme principale de l'énolase, qui compte pour plus de 90 % de l'activité cellulaire de l'énolase. L'énolase est responsable de la conversion du 2-phosphoglycérate (2-PGA) en phosphoénolpyruvate (PEP) dans la glycolyse. Les cellules cancéreuses pourraient être spécialement sensibilisées à l'inhibition de cette voie métabolique à cause de leur fréquente production majeure d'énergie à partir de la glycolyse, ce qu'on appelle « l'effet Warburg ». Le silencement de l'ENO2 par un ARNsh inhibe la croissance, la survie et le potentiel cancérigène des lignées cellulaires où ENO1 est silencé (« ENO1-null ») par rapport à celles où ENO1 est intact (« ENO1-intact »). De plus, l'inhibiteur de l'énolase PhAH, avec des concentrations de demi-inhibition maximales (IC50) autour de 20 nM, a prouvé qu'il était sélectivement toxique pour les cellules ENO1-null par rapport aux cellules ENO1-intact ou aux astrocytes humains normaux. Bien que cet inhibiteur n'a pas montré d'inhibition sélective de l'isoforme ENO1 sur ENO2, une plus forte toxicité envers les cellules ENO1-null est expliquée par une diminution de l'expression globale de l'énolase dans ces lignées cellulaires, qui retiennent seulement environ 10 % de leur activité énolase normale[1]. SF2312, un antibiotique naturel produit par Micromonospora et actif contre les bactéries en conditions anaérobies, s'est également révélé comme potentiel inhibiteur de l'énolase, avec des concentrations en IC50 allant de 10 à 50 nM. Cet inhibiteur a vu son pouvoir d'inhibition augmenté envers ENO2 par rapport à ENO1 à des concentrations supérieures à son IC50. De plus, une forte toxicité sélective du SF2312 envers plusieurs lignées cellulaires où ENO1 a été supprimé ("ENO1-deleted") a été observée ; ces effets n'ont pas été observés dans les lignées cellulaires où ENO1 a été "sauvé", ont été augmentés à la suite d'un traitement au SF2312 par rapport au traitement au PhAH, et ont été plus "forts" en hypoxie. La mort cellulaire et l'inhibition de la prolifération chez les cellules ENO1-deleted traitées au SF2312 ont été précédées d'une diminution de l'ATP et des autres phosphates à haute énergie, et la conversion du glucose en lactate a été inhibée. Ces effets sont en accord avec les effets de l'inhibition de l'enolase sur la glycolyse, et étaient réversibles par la ré-expression de ENO1 ou la surexpression de ENO2. Ainsi, le ciblage pharmacologique de ENO2 offre des perspectives thérapeutiques prometteuses dans les cancers ayant des délétions de ENO1, grâce à la redondance et la fonction cellulaire essentielle de ces gènes[23].
ME2/Me3
Une fraction considérable (~30 %) des cas d'adénocarcinome ductulaire du pancréas (PDAC) sont caractérisés par une fréquente délétion homozygote du gène suppresseur de tumeurs SMAD4 ainsi qu'une perte concomitante des gènes ménagers environnants (18qso). Parmi ces gènes "passagers" qui sont délétés à cause de leur forte proximité avec SMAD4, l'enzyme malique 2 (gène ME2) a été identifiée comme conférant une létalité collatérale, avec ME3 comme partenaire de létalité collatérale. ME2 et ME3 sont des décarboxylases oxydatives mitochondriales impliquées dans la conversion catalytique du malate en pyruvate avec la génération concomitante de NAD(P)H. ME1 et ME2 ont des rôles redondants mais importants dans la régénération de la NADPH et de l'homéostasie des ROS. En accord avec le cadre de la létalité collatérale, un silencement par ARNsh de ME3 dans une gamme de lignées cellulaires PDAC résulte seulement dans la mort sélective de cellules PDAC où ME2 a été supprimé mais qui ne sont pas intactes. Le silencement par ARNsh de ME3 dans des cellules ME2-null cause une accumulation de ROS et l'activation de l'AMPK, qui phosphoryle SREBP1 et inhibe son activité et sa translocation nucléaire. SREBP1 régule la transcription du gène BCAT2 qui contrôle le catabolisme de BCAA et la synthèse de la glutamine, qui est cruciale pour la synthèse de nucléotides de novo. En l'absence de deux enzymes maliques mitochondriales s'ensuivent une production réduite de NADPH et une accumulation de ROS, qui rendent une cellule apoptotique. Ensemble, la redondance fonctionnelle et l'essentialité cellulaire dans l'expression des gènes de létalité collatérale ME2 et ME3 laissent entrevoir une stratégie thérapeutique prometteuse dans le traitement de patients PDAC qui peut être mise en place en ciblant pharmacologiquement le paralogue redondant ME3 du gène "passager" supprimé ME2[24].
TP53/POLR2A
La plupart des cancers humains sont caractérisés par une inactivation du gène suppresseur de tumeur TP53 qui encode la protéine p53, soit par mutation soit par perte du gène. La délétion génomique du locus 17p13.1 de TP53 implique également la co-délétion de gènes environnants comme POL2RA. Ce dernier encode la sous-unité catalytique du complexe de l'ARN polymérase II et est sensible à l'inhibition par l'alpha-amanitine. Dans le cancer colorectal avec inhibition hémizygote de TP53 et POLR2A, l'inhibition de POLR2A amène à une inhibition sélective de la prolifération des cellules cancéreuses indépendamment de la p53. L'inhibition de la POLR2A dans les cellules où celui-ci est intacte n'a aucun effet significatif sur la prolifération cellulaire, la survie et l'apparition de tumeurs, mais cette inhibition est en revanche dramatique sur les cellules hémizygotes pour POL2RA. Cela suggère que la vulnérabilité collatérale et thérapeutique exposée par la délétion hémizygote de TP53 et POL2RA dans le cancer colorectal peut être exploitée pour traiter les cancers colorectaux avec TP53 supprimé en utilisant des inhibiteurs de POL2RA[25].
Déficiences dans le système DDR
Déficience dans le mismatch repair
Des mutations dans les gènes employés lors du mismatch repair (MMR) causent un fort taux de mutations[26] - [27]. Dans les tumeurs, de telles mutations sous-jacentes fréquentes génèrent souvent des antigènes immunogéniques du "non-soi". Un essai clinique Phase II humain réalisé sur 41 patients a évalué une approche de létalité synthétique pour des tumeurs avec ou sans déficiences dans le MMR[28]. Dans le cas des tumeurs sporadiques évaluées, la majorité serait déficiente dans le MMR à cause d'une répression épigénétique d'un gène MMR (voir l'article sur le MMR). Le produit du gène PDCD1 (molécule PD-1) réprime usuellement des réponses immunes cytotoxiques. L'inhibition de ce gène permet une meilleure réponse. Dans ce dépistage clinique Phase II réalisé cette fois sur 47 patients, lorsque les patients cancéreux avec une déficience dans le MMR dans leurs tumeurs ont été exposés à un inhibiteur de la molécule PD-1 ; 67 à 78 % de ces patients ont survécu grâce à cette réponse immune. En revanche, pour les patients sans MMR défectueux, l'addition d'un inhibiteur de la PD-1 a fait que seulement 11 % des patients ont survécu grâce à cette réponse immune. Ainsi, l'inhibition de la PD-1 est essentiellement létale synthétique avec des MMR défectueux.
Déficience dans le gène du syndrome de Werner
L'analyse de 630 tumeurs humaines primaires dans 11 tissus montre que l'hyperméthylation du promoteur du gène WRN codant l'hélicase du même nom (avec perte d'expression de la protéine) est un événement courant dans la cancérogénèse[29]. Le promoteur du gène WRN est hyperméthylé dans environ 38 % des cancers colorectaux et des cancers bronchiques non à petites cellules, et dans environ 20 % des cancers de l'estomac, de la prostate, du sein, des lymphomes non hodgkiniens et des chondrosarcomes, ainsi qu'à des niveaux significatifs dans les autres cancers étudiés. L'hélicase WRN est importante dans la réparation de l'ADN par recombinaison homologue et possède également des rôles dans la réparation de l'ADN par jonction d'extrémités non homologues et par excision de bases[30].
Les inhibiteurs de topoisomérase sont fréquemment utilisés dans la chimiothérapie de différents cancers, bien qu'ils causent une suppression de la moelle osseuse, soient cardiotoxiques et possèdent une efficacité variable[31]. Une étude rétrospective datant de 2006, avec un long suivi clinique, a été réalisée sur des patients atteint de cancer du côlon et traités avec de l'irinotécan comme inhibiteur de topoisomérase. Dans cette étude, 45 patients possèdent des promoteurs du gène WRN qui sont hyperméthylés, et 43 autres non[29]. L'irinotécan était plus fortement bénéfique pour les premiers (39,4 mois de survie en moyenne) que pour les seconds (20,7 mois de survie en moyenne). Ainsi, un inhibiteur de topoisomérase s'est révélé être létal synthétique avec une expression défectueuse du gène WRN. Des évaluations approfondies ont aussi indiqué une létalité synthétique d'une expression déficiente du gène WRN et des inhibiteurs de topoisomérase[32] - [33] - [34] - [35] - [36].
Létalité synthétique clinique et pré-clinique de l'inhibiteur de PARP1
Comme passé en revue par Murata et ses collaborateurs[37], cinq différents inhibiteurs de PARP1 sont actuellement testés cliniquement en Phases I, II et III afin de déterminer si des inhibiteurs particuliers sont létaux synthétiques dans une large variété de cancers, dont ceux de la prostate, du pancréas, les tumeurs bronchiques non à petites cellules, les lymphomes, les myélomes multiples et les sarcomes d'Ewing. De plus, dans les études pré-cliniques utilisant des cellules en culture ou chez la souris, des inhibiteurs de PARP1 sont testés pour leur létalité synthétique contre des déficiences épigénétiques et mutationnelles dans environ 20 défauts de réparation de l'ADN au-delà des déficiences dans BRCA1 et 2. Cela inclut des déficiences dans les gènes PALB2, FANCD2, RAD51, ATM, MRE11, TP53, XRCC1 et LSD1.
Létalité synthétique pré-clinique de ARID1A
La molécule ARID1A, un modificateur de chromatine, est requise pour la jonction d'extrémités non homologues, une voie de réparation majeure qui répare les cassures double brin dans l'ADN[38], et qui a également des rôles de régulation de la transcription[39]. Les mutations ARID1A sont l'une des 12 mutations cancérigènes les plus communes[40]. Une mutation ou une expression épigénétique diminuée sur ARID1A[41] a été trouvée dans 17 types de cancer[42]. Des études pré-cliniques sur des cellules et dans des souris a trouvé une létalité synthétique pour une expression défectueuse de ARID1A soit lors de l'inhibition de l'activité méthyltransférase de la protéine EZH2[43] - [44], soit avec l'addition de l'inhibiteur de la kinase dasatinib[45].
Létalité synthétique pré-clinique de RAD52
Il existe deux voies de réparation des cassures double brin par recombinaison homologue. La voie majeure dépend de BRCA1, PALB2 et BRCA2 alors que la voie alternative dépend seulement de RAD52[46]. Des études pré-cliniques impliquant des cellules défectueuses pour BRCA (mutées ou épigénétiquement réduites, en culture ou injectées dans des souris), montrent que l'inhibition de la protéine RAD52 est létale synthétique s'il y a une déficience pour BRCA[47].
Effets secondaires
Bien que des traitements utilisant les propriétés de la létalité synthétique soient capables de stopper ou ralentir la progression des cancers et prolonger l'espérance de vie, chaque traitement létal synthétique possède certains effets secondaires défavorables. Par exemple, plus de 20 % des patients traités avec un inhibiteur de la PD-1 rencontrent des problèmes de fatigue, des éruptions cutanées, du prurit, de la toux, des diarrhées, un appétit diminué, une constipation ou de l'arthralgie[48]. Par conséquent, il est important de déterminer quel défaut est présent dans le système DDR afin que seul un traitement de létalité synthétique efficace soit appliqué, et qu'il ne soumette pas inutilement les patients à des effets secondaires indésirables sans bénéfice direct.
Voir aussi
Notes
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Synthetic lethality » (voir la liste des auteurs).
Références
- Florian L. Muller, Simona Colla, Elisa Aquilanti et Veronica Manzo, « Passenger Deletions Generate Therapeutic Vulnerabilities in Cancer », Nature, vol. 488, no 7411, , p. 337–342 (ISSN 0028-0836, PMID 22895339, PMCID PMC3712624, DOI 10.1038/nature11331, lire en ligne, consulté le )
- Sebastian M.B. Nijman, « Synthetic lethality: General principles, utility and detection using genetic screens in human cells », Febs Letters, vol. 585, no 1, , p. 1–6 (ISSN 0014-5793, PMID 21094158, PMCID PMC3018572, DOI 10.1016/j.febslet.2010.11.024, lire en ligne, consulté le )
- « Redirecting », sur linkinghub.elsevier.com (consulté le )
- (en) Leland H. Hartwell, Philippe Szankasi, Christopher J. Roberts et Andrew W. Murray, « Integrating Genetic Approaches into the Discovery of Anticancer Drugs », Science, vol. 278, no 5340, , p. 1064–1068 (ISSN 0036-8075 et 1095-9203, PMID 9353181, DOI 10.1126/science.278.5340.1064, lire en ligne, consulté le )
- L. Ryan Baugh, Joanne C. Wen, Andrew A. Hill et Donna K. Slonim, « Synthetic lethal analysis of Caenorhabditis elegans posterior embryonic patterning genes identifies conserved genetic interactions », Genome Biology, vol. 6, , R45 (ISSN 1474-760X, DOI 10.1186/gb-2005-6-5-r45, lire en ligne, consulté le )
- (en) John L. Hartman, Barbara Garvik et Lee Hartwell, « Principles for the Buffering of Genetic Variation », Science, vol. 291, no 5506, , p. 1001–1004 (ISSN 0036-8075 et 1095-9203, PMID 11232561, DOI 10.1126/science.1056072, lire en ligne, consulté le )
- « Synthetic lethal mutations », sur www.sci.sdsu.edu (consulté le )
- Nidhi Saini, Oleg Georgiev et Walter Schaffner, « The parkin Mutant Phenotype in the Fly Is Largely Rescued by Metal-Responsive Transcription Factor (MTF-1) ▿ », Molecular and Cellular Biology, vol. 31, no 10, , p. 2151–2161 (ISSN 0270-7306, PMID 21383066, PMCID PMC3133352, DOI 10.1128/MCB.05207-11, lire en ligne, consulté le )
- (en) Renata Matuo, Fabrício G. Sousa, Daniele G. Soares et Diego Bonatto, « Saccharomyces cerevisiae as a model system to study the response to anticancer agents », Cancer Chemotherapy and Pharmacology, vol. 70, no 4, , p. 491–502 (ISSN 0344-5704 et 1432-0843, DOI 10.1007/s00280-012-1937-4, lire en ligne, consulté le )
- (en) Michael Costanzo, Anastasia Baryshnikova, Jeremy Bellay et Yungil Kim, « The Genetic Landscape of a Cell », Science, vol. 327, no 5964, , p. 425–431 (ISSN 0036-8075 et 1095-9203, PMID 20093466, DOI 10.1126/science.1180823, lire en ligne, consulté le )
- (en) Douglas B. Wheeler, Steve N. Bailey, David A. Guertin et Anne E. Carpenter, « RNAi living-cell microarrays for loss-of-function screens in Drosophila melanogaster cells », Nature Methods, vol. 1, no 2, , p. 127–132 (ISSN 1548-7091, DOI 10.1038/nmeth711, lire en ligne, consulté le )
- (en) Hannah Farmer, Nuala McCabe, Christopher J. Lord et Andrew N. J. Tutt, « Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy », Nature, vol. 434, no 7035, , p. 917–921 (ISSN 0028-0836, DOI 10.1038/nature03445, lire en ligne, consulté le )
- Zhenyu Ji, Fang C. Mei, Pedro L. Lory et Scott R. Gilbertson, « Chemical genetic screening of KRAS-based synthetic lethal inhibitors for pancreatic cancer », Frontiers in bioscience : a journal and virtual library, vol. 14, , p. 2904–2910 (ISSN 1093-9946, PMID 19273243, PMCID PMC2654594, lire en ligne, consulté le )
- Dan Gao, James G. Herman et Mingzhou Guo, « The clinical value of aberrant epigenetic changes of DNA damage repair genes in human cancer », Oncotarget, vol. 7, no 24, , p. 37331–37346 (ISSN 1949-2553, PMID 26967246, PMCID PMC5095080, DOI 10.18632/oncotarget.7949, lire en ligne, consulté le )
- (en) Michael B. Kastan, « DNA Damage Responses: Mechanisms and Roles in Human Disease », Molecular Cancer Research, vol. 6, no 4, , p. 517–524 (ISSN 1541-7786 et 1557-3125, PMID 18403632, DOI 10.1158/1541-7786.MCR-08-0020, lire en ligne, consulté le )
- (en) Carol Bernstein, Anil R., Valentine Nfonsam et Harris Bernstei, DNA Damage, DNA Repair and Cancer, InTech, (DOI 10.5772/53919, lire en ligne)
- Heather M. O'Hagan, Helai P. Mohammad et Stephen B. Baylin, « Double Strand Breaks Can Initiate Gene Silencing and SIRT1-Dependent Onset of DNA Methylation in an Exogenous Promoter CpG Island », PLoS Genetics, vol. 4, no 8, (ISSN 1553-7390, PMID 18704159, PMCID PMC2491723, DOI 10.1371/journal.pgen.1000155, lire en ligne, consulté le )
- Concetta Cuozzo, Antonio Porcellini, Tiziana Angrisano et Annalisa Morano, « DNA Damage, Homology-Directed Repair, and DNA Methylation », PLoS Genetics, vol. 3, no 7, (ISSN 1553-7390, PMID 17616978, PMCID PMC1913100, DOI 10.1371/journal.pgen.0030110, lire en ligne, consulté le )
- Bert Vogelstein, Nickolas Papadopoulos, Victor E. Velculescu et Shibin Zhou, « Cancer Genome Landscapes », Science (New York, N.Y.), vol. 339, no 6127, , p. 1546–1558 (ISSN 0036-8075, PMID 23539594, PMCID PMC3749880, DOI 10.1126/science.1235122, lire en ligne, consulté le )
- Shawn E. Yost, Erin N. Smith, Richard B. Schwab et Lei Bao, « Identification of high-confidence somatic mutations in whole genome sequence of formalin-fixed breast cancer specimens », Nucleic Acids Research, vol. 40, no 14, , e107 (ISSN 0305-1048, PMID 22492626, PMCID PMC3413110, DOI 10.1093/nar/gks299, lire en ligne, consulté le )
- Michael F. Berger, Eran Hodis, Timothy P. Heffernan et Yonathan Lissanu Deribe, « Melanoma genome sequencing reveals frequent PREX2 mutations », Nature, vol. 485, no 7399, , p. 502–506 (ISSN 0028-0836, PMID 22622578, PMCID PMC3367798, DOI 10.1038/nature11071, lire en ligne, consulté le )
- (en) Carol Bernstein, Anil R., Valentine Nfonsam et Harris Bernstei, DNA Damage, DNA Repair and Cancer, InTech, (DOI 10.5772/53919, lire en ligne)
- Paul G. Leonard, Nikunj Satani, David Maxwell et Yu-Hsi Lin, « SF2312 is a natural phosphonate inhibitor of Enolase », Nature chemical biology, vol. 12, no 12, , p. 1053–1058 (ISSN 1552-4450, PMID 27723749, PMCID PMC5110371, DOI 10.1038/nchembio.2195, lire en ligne, consulté le )
- Prasenjit Dey, Joelle Baddour, Florian Muller et Chia Chin Wu, « Genomic deletion of malic enzyme 2 confers collateral lethality in pancreatic cancer », Nature, vol. 542, no 7639, , p. 119–123 (DOI 10.1038/nature21052, lire en ligne)
- Yunhua Liu, Xinna Zhang, Cecil Han et Guohui Wan, « TP53 loss creates therapeutic vulnerability in colorectal cancer », Nature, vol. 520, no 7549, , p. 697–701 (ISSN 0028-0836, PMID 25901683, PMCID PMC4417759, DOI 10.1038/nature14418, lire en ligne, consulté le )
- Latha Narayanan, James A. Fritzell, Sean M. Baker et R. Michael Liskay, « Elevated levels of mutation in multiple tissues of mice deficient in the DNA mismatch repair gene Pms2 », Proceedings of the National Academy of Sciences of the United States of America, vol. 94, no 7, , p. 3122–3127 (ISSN 0027-8424, PMID 9096356, lire en ligne, consulté le )
- Denise Campisi Hegan, Latha Narayanan, Frank R. Jirik et Winfried Edelmann, « Differing patterns of genetic instability in mice deficient in the mismatch repair genes Pms2, Mlh1, Msh2, Msh3 and Msh6 », Carcinogenesis, vol. 27, no 12, , p. 2402–2408 (ISSN 0143-3334, PMID 16728433, PMCID PMC2612936, DOI 10.1093/carcin/bgl079, lire en ligne, consulté le )
- D.T. Le, J.N. Uram, H. Wang et B.R. Bartlett, « PD-1 Blockade in Tumors with Mismatch-Repair Deficiency », The New England journal of medicine, vol. 372, no 26, , p. 2509–2520 (ISSN 0028-4793, PMID 26028255, PMCID PMC4481136, DOI 10.1056/NEJMoa1500596, lire en ligne, consulté le )
- Ruben Agrelo, Wen-Hsing Cheng, Fernando Setien et Santiago Ropero, « Epigenetic inactivation of the premature aging Werner syndrome gene in human cancer », Proceedings of the National Academy of Sciences of the United States of America, vol. 103, no 23, , p. 8822–8827 (ISSN 0027-8424, PMID 16723399, PMCID PMC1466544, DOI 10.1073/pnas.0600645103, lire en ligne, consulté le )
- Raymond J. Monnat, « Human RECQ helicases: Roles in DNA metabolism, mutagenesis and cancer biology », Seminars in cancer biology, vol. 20, no 5, , p. 329–339 (ISSN 1044-579X, PMID 20934517, PMCID PMC3040982, DOI 10.1016/j.semcancer.2010.10.002, lire en ligne, consulté le )
- Yves Pommier, « Drugging Topoisomerases: Lessons and Challenges », ACS chemical biology, vol. 8, no 1, , p. 82–95 (ISSN 1554-8929, PMID 23259582, PMCID PMC3549721, DOI 10.1021/cb300648v, lire en ligne, consulté le )
- Lin Wang, Li Xie, Jun Wang et Jie Shen, « Correlation between the methylation of SULF2 and WRN promoter and the irinotecan chemosensitivity in gastric cancer », BMC Gastroenterology, vol. 13, , p. 173 (ISSN 1471-230X, PMID 24359226, PMCID PMC3877991, DOI 10.1186/1471-230X-13-173, lire en ligne, consulté le )
- (en) Joseph L. E. Bird, Katrin C. B. Jennert-Burston, Marcus A. Bachler et Penelope A. Mason, « Recapitulation of Werner syndrome sensitivity to camptothecin by limited knockdown of the WRN helicase/exonuclease », Biogerontology, vol. 13, no 1, , p. 49–62 (ISSN 1389-5729 et 1573-6768, DOI 10.1007/s10522-011-9341-8, lire en ligne, consulté le )
- KENTA MASUDA, KOUJI BANNO, MEGUMI YANOKURA et KOSUKE TSUJI, « Association of epigenetic inactivation of the WRN gene with anticancer drug sensitivity in cervical cancer cells », Oncology Reports, vol. 28, no 4, , p. 1146–1152 (ISSN 1021-335X, PMID 22797812, PMCID PMC3583574, DOI 10.3892/or.2012.1912, lire en ligne, consulté le )
- Kazunobu Futami, Motoki Takagi, Akira Shimamoto et Masanobu Sugimoto, « Increased Chemotherapeutic Activity of Camptothecin in Cancer Cells by siRNA-Induced Silencing of WRN Helicase », Biological and Pharmaceutical Bulletin, vol. 30, no 10, , p. 1958–1961 (DOI 10.1248/bpb.30.1958, lire en ligne, consulté le )
- (en) Kazunobu Futami, Yuichi Ishikawa, Makoto Goto et Yasuhiro Furuichi, « Role of Werner syndrome gene product helicase in carcinogenesis and in resistance to genotoxins by cancer cells », Cancer Science, vol. 99, no 5, , p. 843–848 (ISSN 1349-7006, DOI 10.1111/j.1349-7006.2008.00778.x, lire en ligne, consulté le )
- Stephen Murata, Catherine Zhang, Nathan Finch et Kevin Zhang, « Predictors and Modulators of Synthetic Lethality: An Update on PARP Inhibitors and Personalized Medicine », BioMed Research International, vol. 2016, (ISSN 2314-6133, PMID 27642590, PMCID PMC5013223, DOI 10.1155/2016/2346585, lire en ligne, consulté le )
- (en) Reiko Watanabe, Ayako Ui, Shin-ichiro Kanno et Hideaki Ogiwara, « SWI/SNF Factors Required for Cellular Resistance to DNA Damage Include ARID1A and ARID1B and Show Interdependent Protein Stability », Cancer Research, vol. 74, no 9, , p. 2465–2475 (ISSN 0008-5472 et 1538-7445, PMID 24788099, DOI 10.1158/0008-5472.CAN-13-3608, lire en ligne, consulté le )
- Jesse R. Raab, Samuel Resnick et Terry Magnuson, « Genome-Wide Transcriptional Regulation Mediated by Biochemically Distinct SWI/SNF Complexes », PLoS Genetics, vol. 11, no 12, (ISSN 1553-7390, PMID 26716708, PMCID PMC4699898, DOI 10.1371/journal.pgen.1005748, lire en ligne, consulté le )
- Michael S. Lawrence, Petar Stojanov, Craig H. Mermel et Levi A. Garraway, « Discovery and saturation analysis of cancer genes across 21 tumor types », Nature, vol. 505, no 7484, , p. 495–501 (ISSN 0028-0836, PMID 24390350, PMCID PMC4048962, DOI 10.1038/nature12912, lire en ligne, consulté le )
- Xianyu Zhang, Qian Sun, Ming Shan et Ming Niu, « Promoter Hypermethylation of ARID1A Gene Is Responsible for Its Low mRNA Expression in Many Invasive Breast Cancers », PLoS ONE, vol. 8, no 1, (ISSN 1932-6203, PMID 23349767, PMCID PMC3549982, DOI 10.1371/journal.pone.0053931, lire en ligne, consulté le )
- Jennifer N. Wu et Charles W. M. Roberts, « ARID1A Mutations in Cancer: Another Epigenetic Tumor Suppressor? », Cancer discovery, vol. 3, no 1, , p. 35–43 (ISSN 2159-8274, PMID 23208470, PMCID PMC3546152, DOI 10.1158/2159-8290.CD-12-0361, lire en ligne, consulté le )
- Benjamin G. Biter, Katherine M. Aird, Azat Garipov et Hua Li, « Targeting EZH2 methyltransferase activity in ARID1A mutated cancer cells is synthetic lethal », Nature medicine, vol. 21, no 3, , p. 231–238 (ISSN 1078-8956, PMID 25686104, PMCID PMC4352133, DOI 10.1038/nm.3799, lire en ligne, consulté le )
- Kimberly H. Kim, Woojin Kim, Thomas P. Howard et Francisca Vazquez, « SWI/SNF mutant cancers depend upon catalytic and non–catalytic activity of EZH2 », Nature medicine, vol. 21, no 12, , p. 1491–1496 (ISSN 1078-8956, PMID 26552009, PMCID PMC4886303, DOI 10.1038/nm.3968, lire en ligne, consulté le )
- (en) Rowan E. Miller, Rachel Brough, Ilirjana Bajrami et Chris T. Williamson, « Synthetic Lethal Targeting of ARID1A-Mutant Ovarian Clear Cell Tumors with Dasatinib », Molecular Cancer Therapeutics, vol. 15, no 7, , p. 1472–1484 (ISSN 1535-7163 et 1538-8514, PMID 27364904, DOI 10.1158/1535-7163.MCT-15-0554, lire en ligne, consulté le )
- B H Lok, A C Carley, B Tchang et S N Powell, « RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination », Oncogene, vol. 32, no 30, , p. 3552–3558 (DOI 10.1038/onc.2012.391, lire en ligne)
- Kimberly Cramer-Morales, Margaret Nieborowska-Skorska, Kara Scheibner et Michelle Padget, « Personalized synthetic lethality induced by targeting RAD52 in leukemias identified by gene mutation and expression profile », Blood, vol. 122, no 7, , p. 1293–1304 (ISSN 0006-4971, PMID 23836560, PMCID PMC3744994, DOI 10.1182/blood-2013-05-501072, lire en ligne, consulté le )
- Jeryl Villadolid et Asim Amin, « Immune checkpoint inhibitors in clinical practice: update on management of immune-related toxicities », Translational Lung Cancer Research, vol. 4, no 5, , p. 560–575 (ISSN 2218-6751, PMID 26629425, PMCID PMC4630514, DOI 10.3978/j.issn.2218-6751.2015.06.06, lire en ligne, consulté le )