Glioblastome multiforme
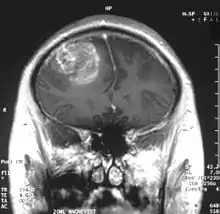
Le glioblastome multiforme (GBM) ou glioblastome, également connu sous le nom d'« astrocytome de grade 4 », est la tumeur primitive du cerveau la plus fréquente et la plus agressive.
| Médicament | Témozolomide et 4,4'-(1,3-propanediylbis(oxy))bis-benzoic acid (d) |
|---|---|
| Spécialité | Neuro-oncologie |
| CIM-10 | C71 |
|---|---|
| CIM-9 | 191 |
| ICD-O | M9440/3 |
| OMIM | 137800 |
| DiseasesDB | 29448 |
| eMedicine |
1156220 med/2692 |
| MeSH | D005909 |
![]() Mise en garde médicale
Mise en garde médicale

.jpg.webp)
Le traitement peut comprendre de la chimiothérapie, de la radiothérapie et de la chirurgie. Ces mesures sont considérées comme palliatives, c'est-à-dire qu'elles ne permettent pas la guérison. L'espérance de vie à cinq ans de cette maladie a peu évolué ces trente dernières années, et ne dépasse pas les dix pour cent. Même avec une résection chirurgicale complète de la tumeur, combinée aux meilleurs traitements disponibles, le taux de survie au GBM reste très faible.
Épidémiologie
Le glioblastome multiforme (GBM) représente 70 % des tumeurs primitives malignes du cerveau aux États-Unis[1] et 20 % de toutes les tumeurs intracrâniennes. Le GBM reste rare et ne représente que 2 ou 3 cas pour 100 000 personnes en Europe et en Amérique du Nord.
Causes
Presque tous les cas de GBM sont sporadiques, sans prédisposition familiale, même si des anomalies chromosomiques somatiques (non constitutionnelles, c'est-à-dire non transmissibles) comme la mutation des gènes PTEN, MDM2, EGFR et P53 sont fréquemment rencontrées dans ces tumeurs. Des signaux anormaux des facteurs de croissance associés aux gènes EGFR, et PDGF (en) sont également perçus. Une délétion du gène NFKBIA (en), codant un inhibiteur du système EGFR, entraîne une surexpression de ce dernier et serait une voie d'activation des glioblastomes[2]. La cause peut être en lien avec un déséquilibre entre des facteurs répresseurs de tumeurs et pro-angiogéniques.
Pathogenèse
Les glioblastomes multiformes sont caractérisés par la présence de petites zones de tissu nécrosé entourées de cellules hautement anaplasiques. Cette caractéristique différencie la tumeur des astrocytomes de grade 3, qui ne possèdent pas ces zones de tissu nécrosé. Bien que le glioblastome multiforme puisse être issu d'astrocytomes de grades inférieurs, des autopsies post-mortem ont révélé que la plupart des glioblastomes multiformes ne sont pas causés par des lésions cérébrales antérieures.
Contrairement aux oligodendrogliomes (en), les glioblastomes multiformes peuvent se former dans la matière grise ou la matière blanche du cerveau, mais la plupart des GBM se forment au plus profond de la matière blanche et infiltrent rapidement le cerveau, devenant parfois très gros avant de donner des symptômes. La tumeur peut s'étendre à la paroi méningée ou à la paroi ventriculaire, entraînant une haute teneur en protéines du liquide cérébrospinal (LCS) (> 100 mg·dL-1) appelée hyperprotéinorachie, et occasionnellement une pléiocytose (en) de 10 à 100 cellules, en majorité des lymphocytes. Les cellules malignes présentes dans le LCR (liquide céphalo-rachidien) peuvent s'étendre à la moelle épinière ou causer une gliomatose méningée. Cependant, les métastases du GBM au-delà du système nerveux central sont extrêmement rares. Environ 50 % des GBM occupent plus d'un lobe d'un hémisphère ou sont bilatéraux. Les tumeurs de ce type naissent généralement dans le cortex, et peuvent traverser le corps calleux, produisant un gliome « papillon » (bilatéral).
La tumeur peut prendre des apparences variées, en fonction de l'étendue de l'hémorragie ou de la nécrose, ou de son âge. Une scanographie (CT scan) montre généralement une masse non homogène avec un centre hypodense étendu en un anneau de taille variable entouré d'œdème. Il existe un effet de masse avec un déplacement possible du ventricule latéral et du troisième ventricule.
Symptômes
Bien que les symptômes communs de la maladie incluent des crises d'épilepsie, des nausées et des vomissements, des maux de tête, et une hémiparésie, le symptôme le plus fréquent est une perte progressive de la mémoire, une dégradation de la personnalité, ou un déficit neurologique dus à un endommagement des lobes frontal et temporal. Le type de symptômes dépend essentiellement de l'emplacement de la tumeur, plus que de ses propriétés pathologiques. La tumeur peut devenir symptomatique rapidement, mais il arrive qu'elle reste asymptomatique malgré une taille énorme.
Diagnostic
Le diagnostic d'un GBM suspecté lors d'un scanner ou une IRM repose sur une biopsie stéréotaxique ou une craniotomie, qui permet, par la même occasion, de retirer autant de tumeur que possible. Bien que la totalité de la tumeur ne puisse théoriquement pas être retirée, en raison de sa multicentricité et de son caractère diffus, une résection partielle peut tout de même prolonger légèrement l'espérance de survie.
Traitement
Le traitement des tumeurs primaires du cerveau et des métastases cérébrales consiste en des thérapies à la fois symptomatiques et palliatives. L'efficacité des traitements reste limitée avec une récidive quasi constante de la tumeur[3]. La taille de la tumeur peut augmenter transitoirement sous traitement (pseudo progression) sans que cela signifie l'échec du traitement[4].
Traitement symptomatique
Le traitement symptomatique consiste à soulager les symptômes et améliorer les fonctions neurologiques du patient. Les principaux médicaments utilisés sont les anticonvulsivants et les corticostéroïdes.
- Les anticonvulsivants sont administrés aux 25 % des patients présentant une épilepsie. Des études ont montré l'inefficacité des anticonvulsivants prophylactiques. Les patients recevant de la phénytoïne parallèlement aux rayons peuvent développer de sérieuses réactions cutanées comme l'érythème multiforme et le syndrome de Stevens-Johnson.
- Les corticostéroïdes, en général la dexaméthasone dont on administre 4 à 10 mg toutes les 4 à 6 heures, peuvent réduire l'œdème péritumoral (par un réarrangement de la barrière hématoméningée), diminuant l'effet de masse et réduisant la pression intracrânienne, entraînant une diminution des maux de tête et de la somnolence.
Traitement antitumoral
Une résection maximale de la tumeur (debulking) est généralement effectuée associée à un protocole de radiothérapie et de chimiothérapie. Cette résection peut être aidé par l'utilisation d'Acide δ-aminolévulinique permettant une accumulation de porphyrines dans les cellules tumorales les rendant fluorescentes[5]. Chez le patient de 70 ans et moins, le traitement de référence depuis 2005 est l'exérèse la plus large possible de la tumeur puis rapidement (entre 2 et 3 semaines et maximum 5 semaines) la mise en place du protocole de Stupp[6]. Le protocole de Stupp est une radio-chimiothérapie concomitante (la radiothérapie est conformationnelle) :
- la radiothérapie est de 2 Gray/jour du lundi au vendredi (repos samedi et dimanche) pendant 6 semaines ; soit 60 Gray en 30 séances ;
- la chimiothérapie est l'agent alkylant : témozolomide TEMODAL. La posologie est de 75 mg/m2/jour pendant 6 semaines (en même temps que la radiothérapie) ;
- puis après un délai d'environ 4 semaines, on entame 6 cycles de témozolomide TEMODAL à la posologie de 150–200 mg/m2/jour de J1 à J5 sur un cycle de 28 jours.
Le témozolomide est surtout efficace en cas de méthylation du gène MGMT[7]. Il peut entrainer des nausées ou des vomissements et un traitement antiémétique par sétrons est souvent proposé. Surveiller le transit intestinal car les sétrons entrainent de la constipation.
Après ces 6 cycles de chimiothérapie seule, un scanner de contrôle est effectué, puis le traitement est proposé au cas par cas. Il n'y a pas de référence, les cycles de chimiothérapie par témozolomide peuvent être continués, une opération peut être réalisée, ou une autre chimiothérapie peut être proposée.
L'application de champs électriques de basse intensité permet d'allonger la durée de rémission[8].
Radiothérapie
Une irradiation crânienne totale (4 500 cGy) avec une dose augmentée (1 500 à 2 000 cGy) à l'emplacement de la tumeur, peut augmenter de 5 mois l'espérance de survie.
La brachythérapie (ou curiethérapie : implantation de grains ou d'aiguilles radioactives) et la radiothérapie ciblée à hautes doses (radiochirurgie stéréotaxique) n'ont montré aucune augmentation de l'espérance de survie.
Chimiothérapie
L'addition de carmustine seule à la radiothérapie augmente légèrement l'espérance de survie. La plupart des oncologues préfèrent une chimiothérapie combinée associant la procarbazine, la lomustine, et la vincristine : le « régime PCV ». Une autre combinaison inclut le carboplatine et le cisplatine. Leur efficacité est limitée et leur toxicité, en particulier avec le régime PCV, est considérable. Le témozolomide a une certaine efficacité[9], surtout chez les patients porteurs d'un variant du gène MGMT[10].
L'ajout de bévacizumab au traitement conventionnel permet un allongement de la durée de vie sans symptômes, sans modifier la survie[11].
La chloroquine[12], médicament antimalarien, a montré une augmentation de la survie à moyen terme lorsqu'elle est combinée à une thérapie conventionnelle (dans ce cas, ablation chirurgicale et traitement à la carmustine).
Autres
Des implants imprégnés de carmustine (Gliadel Wafers) mis en place au moment de la résection primaire ont augmenté la survie moyenne de deux mois[13].
Des espoirs sont fondés sur la thérapie photodynamique (PDT) avec des premiers résultats cliniques[14]. Cette technologie repose sur la combinaison d’un médicament photosensibilisateur et d’un dispositif photonique : lumière laser spécifique et matériel d’illumination adapté à la chirurgie du cerveau.
Côté nutrition, le régime cétogène est à l'étude dans le but d'améliorer la survie des patients. Il a déjà fait ses preuves in vitro et in vivo sur des rongeurs. Cependant, les essais cliniques sur l'homme manquent encore[15].
Traitement palliatif
Un traitement palliatif est fréquemment proposé pour améliorer la qualité de vie du patient dans les dernières semaines.
Récidives
La résurgence de la tumeur après traitement médicamenteux ou chirurgical est quasiment inévitable, en général à moins de 2 cm du site d'origine, et dans 10 % des cas les lésions peuvent apparaître en dehors du site d'origine. La ré-opération ou brachythérapie (curiethérapie) est tentée à nouveau avec des résultats incertains. La thérapie la plus agressive, une seconde intervention et chimiothérapie, est généralement utilisée sur les patients de moins de 40 ans dont la précédente opération remonte à plusieurs mois. Si le régime PCV n'a pas été utilisé, il peut être essayé. Le témozolomide peut être utilisé. Cependant, ces traitements augmentent les intervalles sans aucun symptôme au lieu de prolonger la survie.
Pronostic
La survie médiane sans traitement à partir du diagnostic est de trois mois. Un âge supérieur à 60 ans, une altération de l'état général, une exérèse incomplète sont des facteurs de mauvais pronostic. Le décès est habituellement dû à un œdème cérébral qui entraîne une augmentation de la pression intracrânienne puis une altération de la vigilance.
Avec le traitement standard, c'est-à-dire chirurgie puis radio-chimiothérapie concomitante et chimiothérapie adjuvante avec le témozolomide, la survie médiane est approximativement de 14 mois[16].
Notes et références
- (en) Wen PY, Kesari S, « Malignant gliomas in adults » N Engl J Med. 2008;359:492-507.
- (en) Bredel M, Scholtens DM, Yadav AK, « NFKBIA Deletion in Glioblastomas » N Eng J Med. 2011;364:627-37.
- Schaff LR, Mellinghoff IK, Glioblastoma and Other Primary Brain Malignancies in Adults: A Review, JAMA, 2023;329:574–587
- (en) de Wit MC, de Bruin HG, Eijkenboom W, Sillevis Smitt PA, van den Bent MJ, « Immediate post-radiotherapy changes in malignant glioma can mimic tumor progression », Neurology. 2004;63:535-537.
- Stummer W, Pichlmeier U, Meinel T, Wiestler OD, Zanella F, Reulen HJ; ALA-Glioma Study Group. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: a randomised controlled multicentre phase III trial, Lancet Oncol, 2006;7:392-401
- Stupp R, Mason WP, van den Bent MJ et al; European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma, N Engl J Med. 2005;352:987-996
- Hegi ME, Diserens AC, Gorlia T et al. MGMT gene silencing and benefit from temozolomide in glioblastoma, N Engl J Med, 2005;352:997-1003
- Stupp R, Taillibert S, Kanner A et al. {https://jamanetwork.com/journals/jama/fullarticle/2666504 Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial], JAMA, 2017;318:2306-2316
- (en) Stupp R, Mason WP, van den Bent MJ et al. « Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma » N Engl J Med. 2005;352:987-996.
- (en) Hegi ME, Diserens AC, Gorlia T et al. « MGMT gene silencing and benefit from temozolomide in glioblastoma » N Engl J Med. 2005;352:997-1003.
- (en) Gilbert MR, Dignam J, Won M et al. RTOG 0825: phase III double-blind placebo-controlled trial evaluating bevacizumab (Bev) in patients (Pts) with newly diagnosed glioblastoma (GBM), J Clin Oncol, 2013;31(suppl):1.
- (en) Briceño E, Reyes S, Sotelo J, « Therapy of glioblastoma multiforme improved by the antimutagenic chloroquine », Neurosurg Focus (de), vol. 14, no 2, , e3. (PMID 15727424).
- (en) Westphal M, Hilt DC, Bortey E et al. « A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma » Neuro Oncol (en). 2003;5:79-88.
- (en) Maximilien Vermandel, Clément Dupont, Fabienne Lecomte et Henri-Arthur Leroy, « Standardized intraoperative 5-ALA photodynamic therapy for newly diagnosed glioblastoma patients: a preliminary analysis of the INDYGO clinical trial », Journal of Neuro-Oncology, vol. 152, no 3, , p. 501–514 (ISSN 1573-7373, DOI 10.1007/s11060-021-03718-6, lire en ligne, consulté le )
- (en) Kenneth A. Schwartz, Mary Noel, Michele Nikolai et Howard T. Chang, « Investigating the Ketogenic Diet As Treatment for Primary Aggressive Brain Cancer: Challenges and Lessons Learned », Frontiers in Nutrition, vol. 5, (ISSN 2296-861X, PMID 29536011, PMCID PMC5834833, DOI 10.3389/fnut.2018.00011, lire en ligne, consulté le ).
- (en) Stupp R, Mason W, van den Bent M, Weller M, Fisher B, Taphoorn M, Belanger K, Brandes A, Marosi C, Bogdahn U, Curschmann J, Janzer R, Ludwin S, Gorlia T, Allgeier A, Lacombe D, Cairncross J, Eisenhauer E, Mirimanoff R, « Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma », N Engl J Med, vol. 352, no 10, , p. 987-996 (PMID 15758009).