Aimant monomoléculaire
Un aimant monomoléculaire ou nano-aimant moléculaire, appelé aussi SMM, de l'acronyme anglais Single Molecule Magnet, est une molécule faisant partie des composés de coordination qui a un comportement superparamagnétique: c'est un aimant uniquement en dessous d'une certaine température dite de blocage. Les aimants monomoléculaires sont des macromolécules, c'est-à-dire composés de 100 à 1 000 atomes.
Bien que découverts en 1993, nommés en 1996, l'idée du premier aimant monomoléculaire () fut décrite en 1980. Ils présentent quelques centres magnétiques couplés et isolés de l’environnement extérieur par des ligands volumineux (souvent des ligands organo-carboxylate). Le cœur magnétique est le plus souvent constitué de métaux de transition, les ponts permettant une interaction d'échange entre ces différents constituants sont souvent des composés oxygénés tels que O2−, OH−, OR− ou encore RCO2. Ils donneront naissances aux aimants mono-ioniques, ou Single-Ion Magnets (SIMs) en anglais. Bien que plusieurs centaines d'articles ont été rédigés sur les SMMs et les SIMs, ils ne sont pas encore utilisés industriellement, leur comportement n'apparaissant qu'à de trop faibles températures, en-dessous de la température d'ébullition de l'azote liquide (77 kelvins)[1] - [2].

Ces molécules présentent, en dessous de leur température de blocage, une lente relaxation de l'aimantation d'origine purement moléculaire. Elles peuvent être magnétisées par un champ magnétique extérieur et conserveront cette aimantation même après coupure du champ magnétique. On appelle cela la mémoire magnétique. Par exemple, pour le , après avoir coupé le champ magnétique pendant 4 mois, l'aimantation de la molécule, laissée à 2 kelvins, est toujours présente à 40% de sa valeur de saturation. Si l'on fait le même protocole à 1.5 kelvin, il faudra attendre 40 ans pour obtenir le même résultat (toujours pour le ). Cette propriété est propre à la molécule même, aucune interaction entre molécules n'est nécessaire. L'ordre à longue portée des moments magnétiques n'est pas nécessaire, et le comportement caractéristique du magnétisme moléculaire apparaît même lorsque la molécule est très diluée. C'est une différence notable face aux aimants conventionnels. On peut ainsi dissoudre une SMM dans un solvant diamagnétique et elle montrera toujours cette propriété. Cette lente relaxation de l'aimantation donne lieu à un phénomène d'hystérésis, similaire à ceux observés avec les aimants conventionnels, mais ici d'origine moléculaire : il devient donc possible de stocker de l'information dans une seule molécule[1] - [3] - [4].
Les SMMs combinent les avantages de l'échelle moléculaire, et les propriétés quantiques qui vont avec, aux propriétés magnétiques classiques des aimants macroscopiques conventionnels. Elles possèdent ainsi un grand éventail de propriétés quantiques, allant de l'effet tunnel quantique de l'aimantation à l'interférence de la phase de Berry en passant par la cohérence quantique. Cette dernière étant, grâce à la faiblesse des interactions spin-orbite et hyperfine, bien supérieure à celles habituelles des métaux ou semi-conducteurs. Leurs propriétés magnétiques couplées à leur monodispersité font d'elles des candidates prometteuses au stockage d'information à haute densité, ainsi que, en raison de leur long temps de cohérence, un modèle d'ordinateur quantique[4] - [5].
Historique
Origine du nom
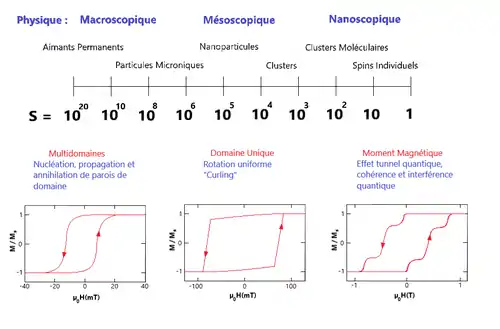
Il existe différentes échelles de taille, allant du macroscopique au nanoscopique, en fonction du nombre de moments magnétiques possédés par la structure. Dans le domaine macroscopique apparaissent les aimants permanents où tous les moments sont bien rangés dans des domaines appelés domaines de Weiss, et séparés par des parois de domaine. Le matériau est alors dit ferromagnétique si tous ces spins magnétiques contribuent positivement à une aimantation nette, ferrimagnétique si une partie des spins se soustrait à l'aimantation nette, ou encore antiferromagnétique si les moments des spins alignés et anti-alignés se compensent complètement. En réduisant maintenant la taille de l'objet, les parois disparaissent progressivement jusqu'à une certaine taille, typiquement de l'ordre d'une dizaine de nanomètres, où la structure est si petite qu'il ne peut plus y avoir de parois. Cette échelle est connue sous le nom de « monodomaine » ou « domaine unique ». Tous les moments magnétiques de la structure sont alors alignés. En réduisant encore la taille du système, passant alors à quelques nanomètres, les moments magnétiques de ce monodomaine deviennent de plus en plus importants et commencent à gouverner les propriétés magnétiques. Si le système est suffisamment petit apparaît un régime quantique. C'est le domaine du spin individuel qui ne peut être décrit qu'au travers de la mécanique quantique[4] - [5] - [6].
Concernant la mécanique du retournement de l'aimantation, à l'échelle macroscopique, l'origine de l'hystérésis magnétique dans un matériel ferromagnétique, c'est-à-dire un matériel multi-domaines, se base sur le mouvement des parois de ces domaines magnétiques (nucléation de domaine, propagation et annihilation de parois de domaine). Lorsque le système est plus petit, de l'ordre du monodomaine, émergent les nanoparticules paramagnétiques. L'origine de leur hystérésis est due à des mécanismes plus simples comme la rotation uniforme du moment magnétique de la particule qui s'aligne avec le champ appliqué, ou plus complexes comme le curling (enroulement de spin). Enfin si le système est suffisamment petit, de l'ordre du nanomètre, des phénomènes quantiques apparaissent comme l'effet tunnel ou les interférences quantiques[4].
C'est ici que les aimants moléculaires trouvent leur place. Dans leur cas, l'hystérésis est due au long temps de relaxation, où, si la température est inférieure à la température de blocage (en-dessous de cette dernière le matériel ne peut changer l'orientation de son moment magnétique, il est alors dit « bloqué »), on observe de brèves relaxations dues à l'effet tunnel. Ce sont d'ailleurs ces brèves relaxations qui provoquent les marches et les sauts dans la courbe d'hystérésis. Le but ici est d'avoir un domaine magnétique suffisamment petit pour que des phénomènes quantiques puissent entrer en jeu. Il a donc été choisi de prendre comme taille celle d'une molécule, soit quelques ångströms. D'où le nom d'aimant mono-moléculaire. Le comportement magnétique de l'aimant se trouve dans une seule molécule et est, comme dit dans l'introduction, isolé de tout environnement extérieur. Néanmoins cela n'est pas entièrement vrai puisqu'il a été prouvé qu'il est nécessaire que la molécule possède son champ de ligand afin d'avoir une lente relaxation de l'aimantation. Ceci est encore plus vrai pour les SIMs. Ces derniers, que l'on peut nommer aimants mono-ioniques en français, sont une sous-division des SMMs dans laquelle le terme de spin électronique émane d'un seul centre magnétique. Son nom induit encore plus en erreur dans le sens où l'on pourrait croire que seul l'ion est nécessaire au comportement magnétique. Or ici aussi le champ de ligand est une condition vitale à la lente relaxation de l'aimantation. Tous les SIMs sont en réalité moléculaires[5] - [7].
Les premiers aimants monomoléculaires
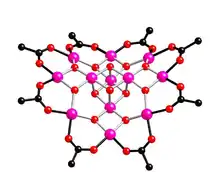
Les premiers SMMs, découverts en 1993, sont devenus les exemples les plus connus. Ils seront plus loin dans l'article utilisés en tant qu'exemples importants afin d'expliquer le comportement des SMMs. Ce sont les composés du manganèse [ Mn12 O12 (O2CCH3)16 (H2O)4 ]·4 (H2O)·2 (CH3CO2H) et du fer [Fe8 O2 (OH)12 (tacn)6]8+ où tacn représente le composé 1,4,7-triazacyclononane, plus connus sous les noms de [8] et . Ces deux exemples sont souvent utilisés afin d'expliquer le phénomène de lente relaxation de l'aimantation[1] - [4] - [5] - [9] - [10]. Pour qu'une molécule magnétique se comporte comme un nano-aimant, elle doit avoir une forte anisotropie magnétique et un grand état fondamental de spin. Ces deux molécules en sont d'excellents exemples. Par résonance paramagnétique électronique à haute fréquence (HF-EPR), on trouve qu'elles ont toutes deux un grand spin total . La molécule possède un noyau avec une structure de type cubique, entouré de huit autres Mn (III) disposés en forme d'anneau. Huit oxoanions et seize ligands acétates viennent compléter la coordination. Les quatre Mn (IV) possèdent une configuration électronique , ainsi chacun contribue avec un spin . Les huit Mn (III) ont eux une configuration et donc un spin . La forte interaction antiferromagnétique entre ces deux parties résulte bien en un état fondamental de spin total . Dans le cas de la molécule , les deux ions fer (III) internes ont une coordinence octaédrique avec les deux oxydes et les quatre ponts hydroxy. Les 6 ions Fe (III) externes s'organisent autour de trois azotes et trois hydroxyles. Chaque ion Fe a un spin 5/2, six d'entre eux ont un spin up et les deux autres un down. Par couplage superéchange on a bien un état fondamental de spin total , ce qui, par éclatement du multiplet fondamental (Zero-Field Splitting), leur font à chacune 21 états que l'on numérote à l'aide d'un nombre quantique , entier compris entre et , et ayant une énergie .












Les SIMs
Les Single-Ion Magnets (SIMs) ou, comme il est possible de les appeler en français, aimants mono-ioniques, sont simplement une sous-division des SMMs, dans laquelle le terme de spin électronique émane d'un seul centre magnétique. L'ion portant le spin est le plus souvent un métal de transition, un lanthanide[11] ou un actinide. Comme vu précédemment, un champ de ligand est nécessaire à la lente relaxation de l'aimantation. De fait, cette dernière n'est observée que lorsque l'ion métallique est placé dans un champ de ligands prévenant de la dégénérescence orbitalaire. Ils sont uniquement nommés ainsi afin de marquer une différence avec les complexes contenant plusieurs centres de spin, où les interactions d'échanges influent le comportement magnétique. Depuis les 15 dernières années, le champ du magnétisme moléculaire a subi un changement d'intérêt, passant des complexes polynucléaires aux SIMs, et en particulier ceux contenant des éléments du bloc f du tableau périodique. Comme dit plus haut, pour avoir des SMMs à haute température il y a deux paramètres définissant la relaxation à maximiser : le spin total et le coefficient d'anisotropie longitudinale . Néanmoins ces deux paramètres ne sont pas facilement manipulables, et bien souvent un fort spin total implique une forte isotropie. C'est en 2003 qu'une approche prometteuse et fondamentalement différente fut proposée par Ishikawa et ses collègues, qui démontrèrent qu'une lente relaxation magnétique peut apparaître dans des complexes lanthanides monomoléculaires. Les plus connus étant ceux où un ion lanthanide est pris en sandwich entre deux ligands de phtalocyanine. La grande différence entre un ion magnétique, comme ce complexe mononucléaire de lanthanide, et une molécule magnétique, comme le cluster étudié plus haut, est que l'ion lanthanide, étant un métal lourd, présente une forte constante spin-orbite mono-électronique, ce qui revient à dire qu'il possède une forte interaction spin-orbite. Il devient donc nécessaire de considérer la valeur du moment cinétique total J (souvent appelé « moment angulaire » par anglicisme) afin de déterminer le niveau fondamental, tandis que, en ce qui concerne les SMMs, le niveau fondamental était déterminé par le moment intrinsèque de spin . De plus, l'anisotropie magnétique provient, dans le cas de l'ion lanthanide, de l'interaction spin-orbite et non pas du champ cristallin. Ce résultat marque le début d'une nouvelle ère dans la détermination des facteurs de la dynamique de spin des SIMs de lanthanides. Des modèles ont été utilisés afin de prédire quel environnement de ligands permet la meilleure optimisation du temps de relaxation, afin de fabriquer des dispositifs moléculaires pouvant stocker et traiter de l'information au-dessus de températures cryogéniques[12] - [13].


Un progrès historique important : le dysprosocenium
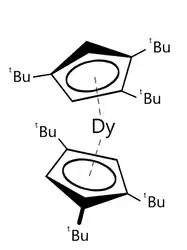
Dans les 25 années qui ont suivi la découverte des aimants monomoléculaires, la température d'hystérésis est passée de 4 kelvins à seulement 14 kelvins environ, en utilisant une vitesse de balayage du champ magnétique constante d'environ 20 œrsteds par seconde. Des températures plus hautes ont été observées en utilisant des vitesses bien plus rapides (30 kelvins ont été atteints à 200 œrsteds par seconde). En , des études ont rapporté un complexe tert-butyle dysprosocenium présentant une température d'hystérésis magnétique de 60 kelvins à une vitesse de balayage de 22 œrsteds par seconde. C'est un résultat aussi récent qu'impressionnant, et les raisons expliquant la conservation de son aimantation à si haute température ne sont toujours pas entièrement connues. Ce pourrait être dû à sa structure particulière. C'est le premier exemple de composé de dysprosium (Dy) ayant uniquement des petits cycles aromatiques en contact avec le métal, similairement au ferrocène. C'est une sorte de métallocène un peu penché avec en guise d'élément métallique un atome de dysprosium. Les cinq électrons non appariés formeraient la base de sa mémoire magnétique mais les ligands augmenteraient et stabiliseraient la distribution inégale du moment magnétique, augmentant ainsi sa température de fonctionnement[14] - [15]. On observe un changement significatif dans la dynamique de la relaxation à cette température. Le fait que ce changement de dynamique persiste dans des échantillons magnétiquement dilués porte à croire que l'origine de l’hystérésis magnétique se trouve dans les modes vibrationnels métal-ligand, uniques au dysprosocenium. Des calculs ab initio sur la dynamique de spin démontrent que la relaxation magnétique à haute température est due aux vibrations moléculaires locales.
Ce n'est pas seulement un record mais un véritable bond en avant dans le domaine du magnétisme moléculaire. Ces résultats indiquent que, avec des design moléculaires judicieux, le stockage d'information dans des molécules uniques serait possible à des températures dépassant celle de l'azote liquide. Ainsi ses aimants fonctionneraient par un refroidissement par azote liquide au lieu d'autres gaz liquéfiés plus froids mais surtout bien plus chers et rares.
Le dysprosocenium, de formule où (1,2,4-tri(tert-butyle)cyclopentadiène) et (tert-Butyle) conserve en le record, avec une température d'hystérésis de 60 kelvins[14] - [15].
![{\displaystyle [Dy(Cp^{ttt})_{2}]^{+}}](https://img.franco.wiki/i/7acfd6dca6f0bf03fce5e2e82ae247b2b8dbb365.svg)


Intérêts
Les aimants sont très prisés dans le stockage d'informations, car la très grande majorité des informations des disques durs sont stockées par l'intermédiaire de particules magnétiques. D'où l'intérêt, pour les industriels, d'augmenter la densité d'information stockable, c'est-à-dire augmenter le nombre de bits d'information dans une région donnée. Pour ce faire, la taille de chaque particule magnétique doit être la plus petite possible. Cette taille limite d'un élément de mémoire est fournie par la taille superparamagnétique. En dessous de cette dernière, l'information ne peut être stockée, l'aimantation fluctuant trop librement. Ceci a lieu à température ambiante (soit 300 kelvins) pour des particules d'une taille de l'ordre de plusieurs dizaines de nanomètres. Cependant, de plus petites particules peuvent être utilisées à plus faible température ou en utilisant l'apparition d'effets quantiques. Les SMMs peuvent en effet avoir des propriétés similaires à celles des aimants si on se place à suffisamment basse température. Chaque molécule peut donc être considérée comme une particule magnétique très petite, idéale pour mettre en place de meilleurs dispositifs de stockage d'informations. De plus, afin qu'elles se comportent de la même façon, les particules doivent avoir la même taille, d'où la nécessité d'utiliser des ensembles de particules identiques. En effet, les propriétés des particules magnétiques augmentent exponentiellement avec la taille, ce qui fait qu'une faible différence dans la taille des particules peut induire d'importantes différences de propriétés. C'est un des avantages des SMMs face aux nano-particules composées de métaux, d'alliages métalliques ou d'oxydes métalliques : la taille uniforme. On trouve également d'autres avantages comme la solubilité dans des solvants organiques, ou que leurs ligands périphériques sont facilement modifiables, permettant d'en faire des films minces[16] ou de les attacher à des surfaces ou à des polymères, etc. Le challenge est donc de développer des SMMs à haute température[1].
L'autre défi, allant de pair avec celui-ci, serait de pouvoir faire des mesures magnétiques suffisamment sensibles pour l'échelle d'une molécule (le nanomètre). Il faudrait pouvoir détecter le retournement de spin dans un seul atome, ou molécule, afin de l'utiliser pour stocker de l'information et ainsi augmenter les capacités de stockage. Il faut donc un dispositif répondant aux changements extrêmement fins du champ magnétique. Le plus sensible à l'heure actuelle est le SQUID. Le composant principal du SQUID étant un anneau supraconducteur dont la sensibilité dépend de sa surface, il est nécessaire d'avoir la superficie la plus faible possible. C'est ce qui a amené à l'élaboration des nano-SQUID, où il est également question de diminuer la capacité électrique des jonctions de Josephson[17].
Un autre intérêt serait de les utiliser dans le domaine de la spintronique moléculaire. Cette dernière combine les idées de trois disciplines récentes, la spintronique, l'électronique moléculaire et l'informatique quantique. L'idée est de manipuler les spins et les charges d'appareils électroniques contenant une ou plusieurs molécules. L'avantage principal d'utiliser des molécules organiques est que leurs faibles interactions spin-orbite et hyperfine tendent à préserver une cohérence de spin avec le temps et la distance bien supérieure à celle des métaux ou semi-conducteurs conventionnels. Le principal but est de contrôler intégralement l'initialisation, la manipulation et la lecture des états de spin de la molécule et d'effectuer des opérations quantiques basiques. Plusieurs procédés existent, comme le transistor de spin moléculaire, le spin valve moléculaire, ou encore le dispositif multi-dot moléculaire[5] - [18].
La physique des aimants monomoléculaires
Modèle du spin géant
Les premiers aimants monomoléculaires, et , seront ici utilisés en tant qu'exemple afin d'expliquer la lente relaxation de l'aimantation de ces composés[1] - [4] - [5] - [9]. C'est-à-dire expliquer en détail pourquoi ces composés porte le nom d'aimants.
En principe, un hamiltonien multi-spin peut être obtenu en prenant en compte toutes les interactions d'échanges et les anisotropies magnétiques de chaque ion. Néanmoins, l'espace de Hilbert étant très grand (106) et les constantes de couplage pas très bien connues, le modèle du spin géant est souvent utilisé pour d'écrire de façon efficace l'état fondamental[19]. On obtient ainsi l'hamiltonien suivant :

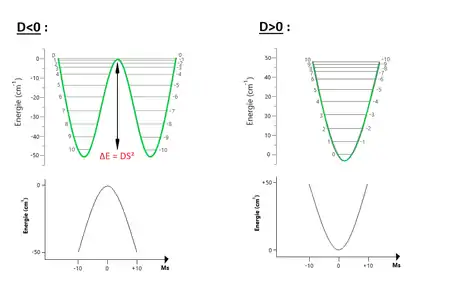
Où , et sont les composantes de l'opérateur de spin, et les constantes d'anisotropie magnétique, mesurée par résonance paramagnétique électronique à haute fréquence (HF-EPR). Les deux premiers termes de cet hamiltonien décrivent de manière effective l'anisotropie magnétique. Cet hamiltonien est dominé par le premier terme qui provient du modèle d'Ising montrant un axe facile suivant , le second terme donne les axes transverses difficiles. Enfin le dernier terme décrit l'énergie Zeeman associée à l'application d'un champ magnétique . Les états de spin excités ainsi que les termes d'anisotropie d'ordres supérieurs sont ici négligés par besoin de simplicité. Dans le cas des SMMs, le terme d'anisotropie axiale est négatif et un axe d'aimantation facile est présent le long de . doit en effet être négatif afin de stabiliser l'état de spin, le modèle d'Ising créant ainsi une barrière énergétique pour le renversement de l'aimantation, c'est-à-dire une barrière entre les spins up et down. En effet à faible température l'aimantation est stabilisée parallèlement ou anti-parallèlement à un axe donné et cette barrière d'énergie devra être surmontée durant son retournement. Comme représenté sur la figure ci-contre, c'est dû au fait que soit négatif que l'état fondamental soit ainsi dégénéré, séparant les spins up et down, et obtenant une bistabilité. Si était positif alors il y aurait un unique état fondamental , plus de barrière énergétique, et la molécule perdrait sa bistabilité.








Ainsi l'énergie potentielle prend la forme d'un double puits[20] (Fig. 3), où les niveaux ont la plus faible énergie, à champ extérieur nul. Afin de renverser l'aimantation, le spin doit surmonter une barrière de potentielle . En dessous apparaissent les tracés des 21 différents états en fonction du nombre quantique . Une parabole est obtenue, inversée ou non suivant le signe de . Ceci est dû au fait que ce soit le premier terme de type Ising qui domine l'hamiltonien. La taille maximale de la barrière est la différence d'énergie entre l'état fondamental et le dernier état excité : , soit une barrière d'environ 67K pour et 25K pour .



Retournement de l'aimantation
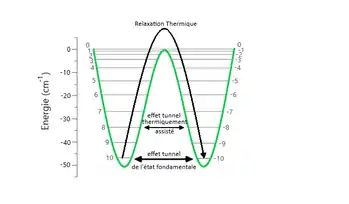
Il existe différentes façons pour le spin de traverser cette barrière énergétique[1] - [2] - [4] - [5] - [10], comme remarqué sur la figure ci-contre (Fig. 4).
La première est la manière classique : par relaxation thermique. À très faible température, quelques dixièmes de kelvin, seul l'état fondamental d'énergie la plus faible est peuplé. Les phonons donnent accès par agitation thermique aux niveaux d'énergies supérieures. Ainsi une température suffisamment grande permet de traverser la barrière classiquement, soit, d'une façon imagée, sauter au-dessus de cette dernière. Or, comme dit précédemment, la taille de la barrière est proportionnelle au carré du spin total . Donc en augmentant le spin total de la molécule, on augmente la taille de la barrière et on rend ainsi impossible un retournement de l'aimantation à plus haute température, ce qui est souhaité pour stocker de l'information. Ce n'est néanmoins pas simple. En effet, augmenter le spin nécessite l'augmentation de la taille des molécules, ce qui est contraignant en soi car une petite taille est préférable afin de stocker un maximum d'information. De plus, l'augmentation de la taille des molécules entraîne une forte augmentation de la symétrie et donc la perte de leur anisotropie[21], caractéristique essentielle au bon fonctionnement des aimants moléculaires.
La seconde est par effet tunnel quantique, et ne dépend pas de la taille de la barrière. En effet, la taille du système est suffisamment petite pour que l'aimantation puisse se renverser par effet tunnel.
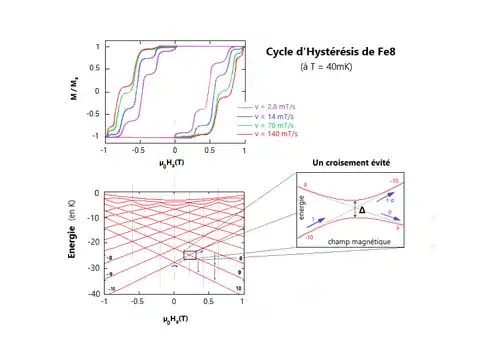
Comme observé sur la figure ci-contre, le cycle d'hystérésis de l'aimantation du composé n'est pas lisse. Des sauts, ou marches, sont observés à intervalles réguliers dans la courbe de l'aimantation en fonction du champ magnétique. Ces marches correspondent à une augmentation brève et importante du renversement de l'aimantation. Telle augmentation a lieu lorsque l'énergie coïncide avec un des niveaux d'énergie de l'autre partie du double puits. Pour ces valeurs d'énergie, l'effet tunnel de l'aimantation est permis, on observe donc une augmentation nette de la vitesse de relaxation, causant ainsi un saut brutal dans la courbe d'hystérésis. On trace les énergies présentes dans la figure n°5 en fonction du champ suivant , c'est-à-dire suivant l'axe facile, et on obtient un diagramme de Zeeman [22] (deuxième graphique de la figure 5). À gauche sont représentés les spins up, dont l'énergie est croissante en fonction du champ, et à droite les down, d'énergie décroissante. On observe des croisements des énergies. Ces croisements sont la cause des sauts soudains du cycle d'hystérésis de l'aimantation, et signifient que le système quantique possède deux états (up et down) avec la même énergie, il est donc dit dégénéré. En réalité, ces énergies ne se croisent pas réellement, on appelle cela des croisements évités, cela signifiant que l'on a une levée de dégénérescence, due aux termes transverses de l'anisotropie magnétique, ou aux champs transverses extérieurs. Dans un croisement évité, l'état de spin a une probabilité de passer par effet tunnel à l'état d'en face et ainsi retourner l'aimantation créant ainsi une marche. Chaque croisement dans le second graphique de la figure n°5 correspond à une marche du premier graphique. Comme cet état a une probabilité de tunnel, il a donc une probabilité de rester dans son état et continuer jusqu'au prochain croisement jusqu'à ce que l'aimantation se soit entièrement retournée. Cette probabilité est donnée par :


![{\displaystyle P(m,m')=1-exp\left[{\frac {-\pi \Delta ^{2}}{4\hbar g\mu _{B}\mu _{0}\mid m-m'\mid dH_{z}/dt}}\right]}](https://img.franco.wiki/i/79552a2a522e147d797f099f1edbd6f9c247369b.svg)
Où est le gap du croisement évité comme montré sur la figure n°5, et les deux nombres quantiques du croisement évité, la constante de Planck, le facteur de Landé (généralement égal à 2), le magnéton de Bohr, la perméabilité magnétique du vide, et enfin la vitesse du balayage. Cette transition non adiabatique entre les deux états d'un système à deux états fut en premier lieu discuté par Landau, Zener et Stückelberg. Elle porte le nom d'effet tunnel Landau-Zener (ou Landau-Zener tunneling en anglais).








Ce modèle permet de comprendre qualitativement le cycle d'hystérésis observé plus haut. Pour un champ magnétique fortement négatif, à très faible température, toutes les molécules sont sur l'état . En augmentant progressivement le champ magnétique, le premier changement apparaît lorsque la région est atteinte, lorsque est proche de zéro. Apparaît alors une probabilité de passer par effet tunnel de l'état à l'état . Il est à noter que plus la vitesse de balayage du champ magnétique est rapide moins la probabilité de tunnel est importante. À l'inverse, plus le gap est grand, plus la probabilité est grande. Ainsi, si est faible alors la probabilité pour le spin de rester sur l'état est très élevée. Le moyen d'échapper à cet état sera lorsque le champ approchera de la région , où il y aura une probabilité d'effet tunnel, permettant de passer à l'état . Comme ce dernier est un état excité, les molécules sur cet état vont se désexciter vers l'état en émettant un phonon. Et ainsi de suite jusqu'à ce que toutes les molécules soient sur l'état . C'est l'inversion de l'aimantation.









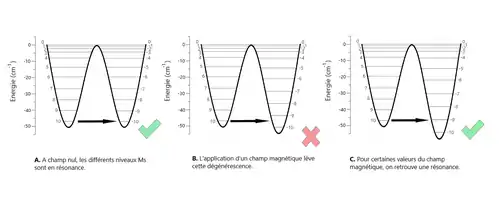
En effet, comme il est possible de le voir sur la figure n° 6, montrant le schéma de l'énergie potentielle d'un aimant moléculaire pour un champ magnétique passant de à , où n est un nombre entier compris entre 1 et 10, l'effet tunnel de l'aimantation n'est présent que lorsque les états sont en résonance, c'est-à-dire lorsque les niveaux d'énergie de chacun des puits sont alignés.


Un champ magnétique appliqué cette fois-ci dans le plan ajuste le splitting à travers les composantes et de l'opérateur de spin des termes Zeeman qui ne commutent pas avec l'hamiltonien de spin. Lorsqu'on trace , où n est un nombre entier compris entre 0 et 10, en fonction du champ magnétique transverse, des oscillations de même fréquence (0.4 T) apparaissent. Une symétrie (ou parité) apparaît selon que n soit paire ou impaire, les maxima des n paires correspondant aux minima des n impaires et vice-versa. Ces oscillations sont expliquées de manière semi-classique par des interférences constructrices ou destructrices des phases quantiques de spin (phase de Berry) de deux chemins tunnels possibles.



Origine de la lente relaxation dans les SIMs
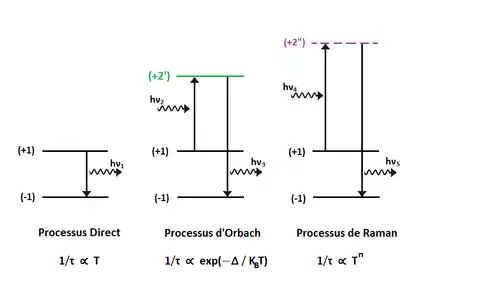
La relaxation de l'aimantation dans les SIMs se produit via différents mécanismes. À faibles températures, c'est-à-dire quelques kelvins, le mécanisme dominant est habituellement l'effet tunnel, régi par des facteurs environnementaux comme la présence de spins nucléaires et de couplages dipolaires. Cet effet tunnel entre états fondamentaux magnétiques est naturellement indépendant de la température. Néanmoins, il est possible pour l'aimantation de passer par effet tunnel aux états excités. Ce processus de relaxation, connu comme effet tunnel thermiquement assisté est quant à lui dépendant de la température. Pour le design des SMMs, l'effet tunnel doit être le plus faible possible.
À des températures plus élevées, le couplage de l'aimantation magnétique aux phonons devenant plus important, d'autres mécanismes de relaxation prennent place. Ces différents processus sont visibles dans la figure n°7, où et sont les deux composantes de la séparation Zeeman, et et symbolisent un état excité réel et virtuel, respectivement. Dans le processus dit direct, la relaxation de l'aimantation de à se fait via l'émission d'un seul phonon d'énergie , où et sont les énergies respectives des états et . Comme peu de phonons correspondent à cette condition, ce processus n'est pas très efficace dans les SIMs. À l'inverse, les processus d'Orbach et de Raman représentées dans la figure 7 sont bien plus performants car ils impliquent des phonons de plus grande énergie. Dans les deux cas, la différence d'énergie entre le phonon absorbé et celui émis est égale à la séparation Zeeman de l'état fondamental : . Les deux processus diffèrent dans les énergies des phonons, et donc dans la nature de l'état excité impliqué dans la relaxation. Alors que dans le cas du processus d'Orbach l'énergie du phonon correspond à l'énergie d'une séparation entre deux états réels, le processus de Raman se fait par absorption et réémission d'un phonon virtuel. On différencie ces processus en caractérisant la relaxation de l'aimantation comme une fonction de la température[12].








Procédés expérimentaux
La lente relaxation de l'aimantation se manifeste comme un cycle d'hystérésis[23]. De fait, le moment magnétique est empêché de se relaxer à sa valeur d'équilibre. La largeur du cycle d'hystérésis augmente généralement lorsque la température diminue. La méthode la plus utilisée pour détecter et caractériser la lente relaxation magnétique se fait par mesure de la susceptibilité magnétique sous courant alternatif (AC) où un faible champ magnétique oscillant est appliqué à l'échantillon. La composante en phase et celle déphasée de la susceptibilité magnétique sont toutes deux mesurées à différentes températures, différentes fréquences d'oscillations et également pour différents champs magnétiques statiques. De toutes ces mesures sont extraits les paramètres de relaxation sous différentes conditions, avec la partie réelle et imaginaire de la susceptibilité magnétique. Ces deux composantes de la susceptibilité sont données par :


- et


où représente le champ magnétique statique, et est le temps de relaxation[24]. Dans le cas simple, selon le modèle de Debye généralisé, le temps de relaxation est associé à l'inverse de la fréquence pour laquelle la susceptibilité déphasé atteint son maximum :




- où


Néanmoins, cette interprétation devient confuse lorsque plus d'un pic est observé. Pour les SIMs, ces différents pics caractéristiques sont liés à des facteurs extrinsèques, comme des interactions dipolaires intermoléculaires[12].
Références
- (en) George Christou, Dante Gatteschi, David N. Hendrickson et Roberta Sessoli, « Single Molecule Magnets Reviews », MRS Bulletin, (DOI 10.1557/mrs2000.226, lire en ligne)
- Olga Iasco, Aimants moléculaires à base de clusters polymétalliques : synthèse, structures cristallines et études des propriétés magnétiques, Université Claude Bernard -Lyon I, (lire en ligne)
- (en) Dante Gatteschi, Roberta Sessoli et Andrea Cornia, « Single molecule magnets based on iron(III) », Royal Society of Chemistry, (DOI 10.1039/A908254I, lire en ligne)
- (en) Wolfgang Wernsdorfer, « Molecular Quantum Spintronics Using Single-Molecule Magnets », Nature Materials, (DOI 10.1038/nmat2133, lire en ligne)
- (en) Wolfgang Wernsdorfer, « Molecular nanomagnets towards molecular spintronics », International Journal of Nanotechnology, (DOI 10.1504/IJNT.2010.031732, lire en ligne)
- (en) Michel Verdaguer et Françoise Villain, How molecules become magnetic ... and the resulting wonderland, Université Pierre-et-Marie-Curie, Paris VI, France (lire en ligne)
- Michel Verdaguer, Anne Bleuzen, Rodrigue Lescouëzec, Valérie Marvaud et Cyrille Train, « (Nano)magnétisme moléculaire », L'Actualité chimique, (lire en ligne)
- (en) Emmanuel Terazzi, Cyril Bourgogne, Richard Welter, Jean-Louis Gallani, Daniel Guillon et Guillaume Rogez, « Single Molecule Magnets with Mesomorphic Lamellar Ordering », Angewandte Chemie International Edition, (DOI 10.1002/anie.20070446, lire en ligne)
- (en) W. Wernsdorfer, S. Bhaduri, D. N. Hendrickson, C. Boskovic et G. Christou, « Spin-parity dependent tunneling of magnetization in single-molecule magnets », American Physical Society (APS), Physical reviews, (DOI 10.1103/PhysRevB.65.180403, lire en ligne)
- (en) Dr. Joris van Slageren, « Introduction to Molecular Magnetism », sur Université de Stuttgart
- (en) Daniel N. Woodruff, Richard E. P. Winpenny et Richard A. Layfield, « Chemical Review : Lanthanide Single-Molecule Magnets », ACS Publication, (DOI 10.1021/cr400018q, lire en ligne)
- (en) Simon G. McAdams, Ana-Maria Ariciu, Andreas K. Kostopoulos, James P.S. Walsh et Floriana Tuna, « Molecular single-ion magnets based on lanthanides and actinides: Design considerations and new advances in the context of quantum technologies », ScienceDirect, (DOI 10.1016/j.ccr.2017.03.015, lire en ligne)
- (en) Shang-Da Jiang, Bing-Wu Wang, Hao-Ling Sun, Zhe-Ming Wang et Song Gao, « An Organometallic Single-Ion Magnet », Journal of the American Chemical Society, (DOI 10.1021/ja200198v, lire en ligne)
- (en) Conrad A. P. Goodwin, Fabrizio Ortu, Daniel Reta, Nicholas F. Chilton et David P. Mills, « Molecular magnetic hysteresis at 60 kelvins in dysprosocenium », Nature, (DOI 10.1038/nature23447, lire en ligne)
- (en) Dr. Fu-Sheng Guo, Dr. Benjamin M. Day, Yan-Cong Chen, Prof. Dr. Ming-Liang Tong, Akseli Mansikkamäki et Prof. Dr. Richard A. Layfield, « A Dysprosium Metallocene Single‐Molecule Magnet Functioning at the Axial Limit », Angewandte Chemie International Edition, (DOI 10.1002/anie.201705426, lire en ligne)
- (en) Massimiliano Cavallini, Massimo Facchini, Cristiano Albonetti et Fabio Biscarini, « Single Molecule Magnets: from thin films to nano-patterns », Physical Chemistry Chemical Physics, (DOI 10.1039/B711677B, lire en ligne)
- (en) R. Russo, E. Esposito, C. Granata, A. Vettoliere, M. Russo, C. Cannas, D. Peddis et D. Fiorani, « Magnetic Nanoparticle Characterization Using Nano-SQUID based on Niobium Dayem Bridges », ScienceDirect, (DOI 10.1016/j.phpro.2012.06.162, lire en ligne)
- (en) ShangDa Jiang, Karin Goß, Christian Cervetti et Lapo Bogani, « An introduction to molecular spintronics », Science China Chemistry, (lire en ligne)
- (en) W. Wernsdorfer, S. Bhaduri, D. N. Hendrickson, R. Tiron et G. Christou, « Spin-Spin Cross Relaxation in Single-Molecule Magnets », American Physical Society (APS), Physical reviews, (DOI 10.1103/PhysRevLett.89.197201, lire en ligne)
- Michel Verdaguer et Cyrille Train, « La molécule, précurseur du solide magnétique », Laboratoire CIMM, (lire en ligne)
- (en) Muralee Murugesu, Malgorzata Habrych, Wolfgang Wernsdorfer, Khalil A. Abboud et George Christou, « Single-Molecule Magnets : A Mn25 Complex with a Record S = 51/2 Spin for a Molecular Species », Journal of the American Chemical Society (JACS), (DOI 10.1021/ja0316824, lire en ligne)
- (en) Wolfgang Wernsdorfer, Núria Aliaga-Alcade, David N. Hendrickson et George Christou, « Exchange-biased quantum tunneling in a supramolecular dimer of single-molecule magnets », Nature, (DOI 10.1038/416406a, lire en ligne)
- (en) En-Che Yang, Nicholas Harden, Wolfgang Wernsdorfer, Lev Zakharov, Euan K. Brechin, Arnold L. Rheingold, George Christou et David N. Hendrickson, « Mn4 Single-Molecule Magnets with a planar diamond core and S=9 », ScienceDirect, (DOI 10.1016/S0277-5387(03)00173-6, lire en ligne)
- (en) Kasper S. Pedersen, Daniel N. Woodruff, Jesper Bendix et Rodolphe Clérac, Lanthanides and Actinides in Molecular Magnetism : Chap.5 : Experimental Aspects of Lanthanide Single-Molecule Magnet, WILEY-VCH, (ISBN 9-783-52733-526-8)
Liens externes
- (en) Several new Single-Molecule Magnets discovered Indiana University
- (en) Lectures notes in Single-Molecule Magnets and nano-magnetism
- (en) National Physical Laboratory (UK), Lecture notes in Single-Molecule Magnets
- (en) Metrological Challenges of Nanomagnetism par Olga Kazakova, Carol Webster et Alexander Tzalenchuk, National Physical Laboratory (UK)
- [vidéo] Molecular Quantum Spintronics using single-molecule magnets par Wolfgang Wernsdorfer, médaillé d'argent CNRS et premier détenteur du prix Olivier Khan sur Youtube
- (en) Chemistry of Nanostructured Materials; Peidong Yuang (UC Berkeley), World Scientific Publishing: Hong Kong, 2003.
- (en) Molecular Magnetism, Olivier Kahn, VHC, New York, 1993.
- (fr) Hommage à Olivier Kahn Magnétisme Moléculaire, coord. Michel Verdaguer, .