Leucémie lymphoïde chronique
La leucémie lymphoïde chronique (LLC) est une maladie cancéreuse du sang (leucémie), caractérisée par la prolifération de lymphocytes ce qui la place dans la catégorie des hémopathies lymphoïdes chroniques. Il s'agit de la leucémie la plus fréquente, touchant de façon préférentielle les personnes âgées de plus de 50 ans. En dehors de quelques cas familiaux, on ne connaît pas de facteurs favorisant le déclenchement de la maladie qu'ils soient environnementaux, génétiques ou infectieux. Cette affection est incurable à ce jour, mais le pronostic est extrêmement variable d'un malade à l'autre. Pour 60 % des patients il n'est pas nécessaire de commencer un traitement lors du diagnostic initial de la maladie compte tenu de sa lente évolutivité.
| Médicament | Uramustine, alemtuzumab, mercaptopurine, (RS)-cyclophosphamide, chlorambucil, pégaspargase (en), cladribine, ifosfamide, phosphate d'étoposide (d), prednisone, bendamustine, (RS)-lénalidomide, pentostatin, nilotinib, pegfilgrastim, doxorubicine, dasatinib monohydraté (d), ibrutinib, idelalisib, dexaméthasone, vénétoclax, vincristine, rituximab, ofatumumab, afutuzumab, chlorambucil, (RS)-cyclophosphamide et vénétoclax |
|---|---|
| Spécialité | Hématologie |
| CISP-2 | B73 |
|---|---|
| CIM-10 | C91.1 |
| CIM-9 | 204.1 |
| ICD-O |
M9823/3 (CLL) 9670/3 (SCL) |
| OMIM | 609630, 109543, 612557, 612559 et 612558 151400, 609630, 109543, 612557, 612559 et 612558 |
| DiseasesDB | 2641 |
| MedlinePlus | 000532 |
| eMedicine | 199313 |
| MeSH | D015451 |
![]() Mise en garde médicale
Mise en garde médicale
Histoire
Les premières recherches sur les leucémies datent du XIXe siècle. La leucémie lymphoïde a été individualisée grâce aux colorations cytologiques d'Ehrlich au sein des leucémies chroniques vers 1880[1].
Physiopathologie et cytologie
La leucémie lymphoïde chronique est définie par la prolifération monoclonale d'une population mature de lymphocytes B (il peut s'agir de lymphocytes de type T dans 5 % des cas) ; ces lymphocytes vont envahir progressivement le sang, les organes lymphoïdes et la moelle osseuse[2].
Les mécanismes cellulaires induisant la prolifération des lymphocytes sont imparfaitement connus. On estime que les lymphocytes en cause dérivent des couronnes péri-folliculaire des ganglions, que certains réarrangements dans les gènes d'immunoglobulines induisent des lymphocytes de type B, que ce sont les processus de mort programmée cellulaire qui sont perturbés et non la surexpression d'oncogène de multiplication. La dysrégulation semble atteindre une sous-population particulière de lymphocytes B étroitement impliquée dans l'ordonnancement de la reconnaissance et la tolérance du soi. Cette atteinte sélective explique probablement la grande quantité de maladies auto-immunes compliquant la leucémie lymphoïde chronique.
L'étude du réarrangement des gènes d'immunoglobulines montre de nombreuses mutations somatiques ce qui est caractéristique des lymphocytes matures à un stade post-germinal, et non pas de lymphocytes naïfs. Il existe également de nombreuses anomalies chromosomiques acquises au sein des lignées leucémiques, ce qui pourrait contribuer à leur prolifération, avec notamment une délétion d'une partie du chromosome 13, une trisomie 12, des délétions sur le chromosome 17 ou 11[3]. Il peut exister des mutations acquises sur certains gènes, comme sur le TP53[4], entraînant une dysfonction de cette voie.
Ces lymphocytes B expriment normalement à leur surface membranaire des protéines antigéniques dites cluster de différenciation (CD) caractéristiques de l'origine B : le CD19, le CD20, ainsi que de façon anormale le CD5 (marqueur exprimé normalement par les lymphocytes T) et le CD23. Les CD5 et CD23 sont parfois présents sur des lymphocytes B lors de leur activation antigénique, mais pas de façon constante. Des lymphocytes B CD5+ se voient également chez des patients atteints de maladies auto-immunes. La population leucémique est ainsi CD19+ CD20+ CD5+.
Les lymphocytes B leucémiques expriment avec une faible intensité la même immunoglobuline de membrane à chaîne légère κ ou λ et chaîne lourde de type M (parfois de type D) ce qui caractérise leur caractère monoclonal (IgG et IgA dans 10 % des cas).
Ces cellules lymphocytaires B sont immuno-incompétentes donc incapables de répondre à la stimulation antigénique pour fabriquer des anticorps et défendre l'organisme contre une agression. Ils expriment de façon réduite, en particulier, les récepteurs membranaires qui sont présents en grand nombre sur les lymphocytes B normaux. Ce déficit ne semble pas être dû à un défaut de synthèse mais à une anomalie de repliement empêchant leur expression sur la membrane cellulaire[5].
Ces lymphocytes B monoclonaux présentent de manière fréquente de nombreuses mutations ainsi que des anomalies chromosomiques sans que le rôle exact de ces derniers dans la genèse de la leucémie puisse être établi[6].
L'hypothèse d'une apoptose réduite (mort cellulaire programmée) a été évoquée comme possible cause de la prolifération de lymphocytes matures : l'expression de certains gènes en rapport avec l'inhibition de l'apoptose semble être augmentée dans certains cas[6].
Causes
Elles sont mal comprises et probablement souvent multifactorielles (peut être, rarement, génétiques car quelques cas familiaux ont été signalés). Comme pour d'autres maladies ayant une composante auto-immune, des facteurs environnementaux sont évoqués, notamment par une étude cas-témoin faite dans 3 régions françaises. Ce travail a recherché les facteurs de risque de développer un lymphome non hodgkinien (hémopathies lymphoïdes telles qu'on les observe depuis quelques décennies). Chez 298 malades, comparés à et 276 témoins) ; les facteurs étudiés ont été la consommation de tabac et d'alcool, l'usage de teintures capillaires, l’exposition au soleil et aux radiographies, l’influence de maladies allergiques, infectieuses et non infectieuses, de médicaments, de vaccins, de la santé et la proximité avec des animaux lors de l'enfance. L'étude (sur la base d'immunophénotypage à partir d'analyses de sang) a décrit des marqueurs de prolifération cellulaire et d’apoptose pour cette maladie, et elle pose la question de l’hypothèse hygiéniste et du « paradigme TH1/TH2 »[7].
Épidémiologie
Épidémiologie descriptive
En matière de leucémie lymphoïde chronique, le ratio homme/femme est de 2/1 et le pic de fréquence se situe vers l'âge de 65 ans. La LLC est exceptionnelle avant 40 ans, touche un patient de plus de 50 ans dans 90 % des cas et de plus de 60 ans dans 66 %.
C'est la leucémie chronique la plus fréquente dans les pays occidentaux, son incidence est évaluée à 3,5 nouveaux cas pour 100 000 personnes par an aux États-Unis[8] avec environ 20 000 nouveaux cas et 4000 décès annuels[9].
Elle est plus rare en Extrême-Orient[6].
Épidémiologie analytique
Il semble parfois exister une prédisposition familiale avec un risque multiplié par trois si un membre de la fratrie est atteint[10], avec des facteurs de confusion parfois possibles.
L'exposition à un pesticide défoliant, l'agent orange (ou aux résidus de Dioxines qu'il contenait), pourrait augmenter le risque de la maladie[6]. Le risque est également majoré chez ceux qui vivent dans une ferme[11].
De même, le rôle potentialisateur d'une irradiation reste controversé[12].
Des infections répétées, en particulier pulmonaires[13], semblent cependant représenter un facteur de risque de la maladie, probablement en raison d'une stimulation itérative du système immunitaire[11].
Certaines essences peuvent également contenir des composés susceptibles de provoquer ce genre de maladie[14].
Éléments diagnostiques
Le diagnostic est souvent suspecté sur l'apparition chez un sujet âgé de nombreux ganglions superficiels et accessibles à la palpation (adénopathies). Le plus souvent le diagnostic est suspecté devant une augmentation du nombre de lymphocytes (lymphocytose) à la numération formule sanguine à plus de 4 000/µl. Dans tous les cas cette augmentation est confirmée sur plusieurs examens.
Présentation clinique
Dans environ un tiers des cas, il n'existe aucun signe clinique de la maladie. Celle-ci est souvent découverte au décours d'une prise de sang (comportant un hémogramme) demandé à titre systématique[3]. La forme clinique la plus fréquente reste la découverte par le patient ou au cours d'un examen d'une adénopathie superficielle ou d'une splénomégalie (rate augmentée de volume). Plus rarement, le diagnostic est porté au détour d'une complication infectieuse ou d'une manifestation auto-immune. La présence d'une fièvre, de sueurs nocturnes ou d'une altération de l'état général doit faire rechercher une infection sous-jacente, une transformation de la LLC en lymphome de haut-grade (syndrome de Richter) ou un cancer touchant d'autres organes.
Bilan systématique
Une fois le diagnostic suspecté, le bilan diagnostique comprendra de façon systématique un hémogramme avec frottis sanguin, une étude immunophénotypique des lymphocytes sanguins par cytométrie en flux, un caryotype, une électrophorèse des protéines sériques complétée par une immunoélectrophorèse, plus sensible, un test de Coombs et une radiographie thoracique.
L'immunophénotypage des lymphocytes sanguins (circulants) est l'examen central du diagnostic. il confirme la présence d'une prolifération monoclonale de lymphocytes B (CD19+ CD20+) co-exprimant CD5+, marqueur habituel des lymphocytes T. Le caractère monoclonal est caractérisé par l'expression d'une seule chaîne légère κ ou λ.
L'immunophénotypage suffit au diagnostic, il permet le calcul du score de Matutes qui établit le diagnostic de LLC s'il est égal à 4 ou 5[15] :
| Points | 1 | 0 |
|---|---|---|
| Immunoglobuline de surface | Expression faible | Expression forte |
| Expression du CD5 | + | - |
| Expression du CD23 | + | - |
| Expression du CD79b | Faible ou nulle | Forte |
| Expression de FMC7 | - | + |
L'hémogramme montrera de façon constante un nombre de lymphocytes supérieure à 4 000/µl (lymphocytose). Cette lymphocytose est indispensable au diagnostic. Elle est variable et atteint parfois des valeurs très élevées (> 200 000/µl). L'anémie et la thrombopénie (baisse du nombre de plaquettes sanguines) ne sont pas systématiques et sont caractéristiques de formes plus graves. Les causes de l'anémie et de la thrombopénie sont plurifactorielles : auto-immunité, érythroblastopénie auto immune, splénomégalie, défaut de production médullaire du fait de l'envahissement de la moelle, et inflammation.
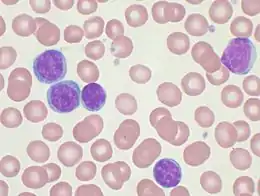
Le frottis sanguin montre fréquemment des cellules altérées et des noyaux nus, appelés « ombre de Gümprecht ».
L'électrophorèse des protides décèle une hypogammaglobulinémie (dans 30 à 60 % des LLC). L'immunoélectrophorèse peut révéler la présence d'une immunoglobulinémie monoclonale (dans 10 % des cas de LLC). Enfin, le test de Coombs peut montrer une auto-immunisation anti-érythrocytaire.
L'examen cytogénétique (caryotype) des cellules de la lignée lymphocytaire n'est pas indispensable au diagnostic mais semble avoir une valeur pronostique importante. Le caryotype retrouve des anomalies chromosomiques dans 50 % à 80 % des cas. Les anomalies les plus fréquemment retrouvées sont une délétion du bras long du chromosome 13 (délétion 13q) et la trisomie du chromosome 12. Certaines anomalies sont de mauvais pronostic comme les délétions des chromosomes 11 et 17 (17p avec perte du gène suppresseur de tumeur P53) de même que des anomalies du bras long du chromosome 11 (11q23).
Bilan optionnel
Depuis le développement de l'immunophénotypage, le myélogramme, nécessitant une ponction de la moelle osseuse, n'est plus indispensable au diagnostic. La biopsie ostéomédullaire ou ganglionnaire peut être utile dans certains diagnostic difficiles ou lors d'une suspicion de transformation en lymphome de haut grade (syndrome de Richter).
Formes cliniques
- Le lymphome lymphocytique ayant un syndrome tumoral au premier plan, prépondérant sur la phase leucémique.
- Les leucémies lymphoïdes chroniques de phénotype B CD5(-) représentent environ 10 % des LLC.
- Les leucémies lymphoïdes chroniques de phénotype T (leucémies à grands lymphocytes granuleux) représentent environ 5 % des LLC.
Une prolifération modérée monoclonale de lymphocytes B (moins de 5000 éléments par mm3, sans signe clinique, est considérée comme une forme précoce de la maladie[16], appelée « lymphocytose B monoclonale »[17]. Son risque évolutif est faible et sa transformation en une leucémie justifiant un traitement est de l'ordre de 1% par an[16].
Diagnostic différentiel
Les principaux diagnostics différentiels de la leucémie lymphoïde chronique peuvent être les hyperlymphocytoses infectieuses, le lymphome non-Hodgkinien, le lymphome du manteau, la leucémie à prolymphocytes, la leucémie à tricholeucocytes, la maladie de Waldenström.
Dans certains cas, il existe un prolifération monoclonale de lymphocytes mais dont le nombre reste en deçà du seuil duquel on pose le diagnostic de LLC. Cette entité, appelé lymphocytose monoclonale de type B pourrait être une forme pré-leucémique, mais cela reste discuté[18].
Histoire naturelle de la maladie
Complications
L'hyperuricémie (augmentation du taux d'acide urique dans le sang) n'est pas rare.
Le syndrome de Richter survient dans 10 % des cas, se caractérisant par un syndrome tumoral lié à l'envahissement de l'organisme par une multiplication de cellules lymphocytaires B pouvant mimer un lymphome malin non-hodgkinien : il s'agit en fait d'un lymphome de type immunoblastique à grandes cellules.
L'insuffisance médullaire peut être consécutive à l'envahissement de la moelle osseuse ce qui vient limiter sa production de cellules sanguines. La diminution de fabrication des globules rouges (érythrocytes) conduit à l'anémie, celle des plaquettes à la thrombopénie et aux syndromes hémorragiques, celle des polynucléaires neutrophiles (granulocytes) favorise les infections bactériennes.
Le déficit d'immunité humoral est en grande partie lié à l'hypogammaglobulinémie qui sera confirmée par un dosage pondéral des IgG, IgM, et IgA. Les infections sont plus fréquentes et touchent la sphère ORL et pulmonaire.
L'auto-immunité qui se développe dans de rares cas peut entraîner des anémies hémolytiques avec test de Coombs direct positif (et baisse de l'haptoglobine).
Pronostic
Le pronostic de la leucémie lymphoïde chronique est extrêmement variable suivant les patients. Des classifications pronostiques ont été développées pour adapter le traitement au rythme évolutif de la maladie. Celles les plus fréquemment utilisées sont la classification de Rai[19] aux États-Unis et la classification de Binet[20] en Europe.
| Classification de Binet[20] | ||||
| Stades | Pronostic | Critères de définition | Répartition des LLC en % | survie médiane (en mois) |
| Stade A | Bon pronostic | Lymphocytose, taux d'hémoglobine > 100 g/l et numération des plaquettes > 100 G/l, moins de trois aires ganglionnaires atteintes |
63 % | > 120 |
| Stade B | Pronostic intermédiaire | Lymphocytose, taux d'hémoglobine > 100 g/l et numération des plaquettes > 100 G/l, plus de trois aires ganglionnaires atteintes |
30 % | 70 |
| Stade C | Mauvais pronostic | Lymphocytose, taux d'hémoglobine < 100 g/l ou numération des plaquettes < 100 G/l, quel que soit le nombre d'aires lymphoïdes atteintes |
7 % | 40 |
Les aires lymphoïdes peuvent être les aires cervicales, axillaires, inguinales (qu'elles soient unilatérales ou bilatérales), la rate ou le foie. Ces anomalies étant déterminées par la palpation lors de l'examen clinique.
| Classification de RAI (publiée en 1975[19] révisée en 1980) | |||
| Stades | Critères de définition | Pronostic | survie médiane (en mois) |
| Stade 0 | Lymphocytose sanguine /L et médullaire isolées sans adénopathies ni splénomégalie | Bon pronostic | > 120 |
| stade I | Stade 0 (hyperleucocytose) et adénopathies sans splénomégalie ni hépatomégalie | pronostic intermédiaire | > 100 |
| Stade II | Stade 0 et splénomégalie et/ou adénopathies et/ou hépatomégalie | 70 | |
| Stade III | Stade 0 et anémie (Hb < 11g/dl) qu'il y ait ou non adénopathies ou splénomégalie ou hépatomégalie | Mauvais pronostic | 24 |
| Stade IV | Stade 0 et thrombopénie (Plaq < 100 000/mm3 ) qu'il y ait ou non adénopathies ou splénomégalie ou hépatomégalie | ||

Ces classifications sont suffisamment précises pour caractériser les formes graves. Il existe cependant de nouveaux marqueurs biologiques permettant de mieux connaître le pronostic des LLC. Certains paramètres biologiques ont une valeur pronostic péjorative comme :
- un temps de doublement du nombre de lymphocytes circulants inférieur à 12 mois ;
- un taux sérique des LDH ou de thymidine kinase[21] élevé ;
- un taux sérique de β2 microglobulines élevé[22] ;
- la présence d'une mutation du TP53[23], ou l'absence de mutation sur le IGHC[24] ;
- une expression du ZAP-70[25], du CD49d[26].
L'ensemble de ces facteurs ont été intégrés dans un nouvel index permettant de prédire les formes à mauvais pronostic[27].
Traitement
La prise en charge de la leucémie lymphoïde chronique a fait l'objet de la publication de recommandations. Celle de l' « International Workshop on Chronic Lymphocytic Leukemia » datent de 2018[28].
Le traitement dépend évidemment du stade de la maladie. Selon la classification de Binet, il n'y a pas d'indication aujourd'hui à traiter les patients au stade A, ce qui correspond à 65 % des patients ; en revanche, il est nécessaire de traiter les patients de stade B ou C. Le traitement repose sur la chimiothérapie orale ou injectable. Les thérapies ciblées tendent à prendre le pas, du moins dans les formes les plus sévères[29]. Dans les formes de mauvais pronostic, il peut être proposé un traitement intensif sous la forme d'une chimiothérapie intensive suivie d'une greffe de moelle; il peut s'agir d'une autogreffe (le donneur étant le patient lui-même), ou d'une allogreffe (le donneur étant un membre de sa fratrie ou un donneur volontaire de moelle).
Les complications évolutives sont traitées de façon spécifique.
Chimiothérapie
Les produits de chimiothérapie utilisés dans le traitement intensif de la leucémie lymphoïde sont le chlorambucil employé seul, la fludarabine employée seule, des cures mensuelles de chimiothérapie de type CHOP (association de quatre agents: Cyclophosphamide-(H)adryamicine-Oncovin(vincristine)-Prednisone).
Thérapie ciblée
Les cellules leucémiques étant CD20+, un anti-CD20 (anticorps monoclonal reconnaissant spécifiquement le CD20) peut être utilisé lors du traitement. Le rituximab (Mabthera), associée à la fludarabine et à la cyclophosphamide fait ainsi partie de l'arsenal thérapeutique[30], constituant la chimiothérapie de référence pour la maladie. Un autre anti CD20, l'obinutuzumab est utilisé avec le chlorambucil[31].
Une autre cible est la tyrosine kinase de Bruton dont l'expression est majorée dans les cellules leucémiques. L'ibrutinib, étant un inhibiteur de cet enzyme, entraînant l'apoptose (la mort) des cellules leucémiques, permet des rémissions plus prolongées, même chez les formes réfractaires ou récidivantes[32]. L'acalabrutinib est également supérieur au traitement conventionnel dans les formes récidivantes ou réfractaires[33].
L'idelalisib a également été testé (il provoque une apoptose) en cas de délétion 17p ou mutation TP53 ; néanmoins, les essais cliniques le concernant ont été arrêtés en raison d'une surmortalité par infections[34]. Le duvelisib, de mode d'action identique, est actif sur les formes réfracatires ou récidivantes[35].
Le venetoclax, un inhibiteur du Bcl-2, permet d'avoir certaines réponses dans des formes réfractaires[36].
Thérapie cellulaire
Le transfert adoptif de cellules ciblant le CD19 constitue une piste de recherche[37].
Traitement des complications auto-immunes
Les complications auto-immunes sont traitées avec une corticothérapie générale continue. En deuxième intention, l'injection en intraveineuse d'immunoglobulines permet une amélioration transitoire. La radiothérapie de la rate voire l'ablation chirurgicale de cette dernière (splénectomie) sont utiles en dernier recours.
Traitement des complications infectieuses
- Les complications infectieuses d'origine bactérienne sont traitées par des antibiotiques adaptés.
- Les infections itératives liées à un déficit de l'immunité humorale caractérisé par une hypogammaglobulinémie peuvent être prévenues par l'utilisation d'immunoglobulines intraveineuses polyvalentes.
- En cas de lymphopénie CD4, traduisant un déficit de l'immunité cellulaire, un traitement préventif de la pneumocystose par Bactrim est indiqué.
Stade A de Binet
Il existe à l'heure actuelle un consensus pour ne pas traiter les patients atteints de LLC au stade A.
Stade B et C de Binet
Le traitement de référence actuel (gold standard) est l'association fludarabine + cyclophosphamide + rituximab[3]. Chez le patient âgé, l'association bendamustine + rituximab, mieux tolérée, peut être proposée[38].
Traitement d'entretien
Différents traitements sont proposés pour minimiser le risque de rechute : l'alemtuzumab[39], l'ofatumumab[40] ou le rituximab[41] parviennent à ce but, avec, cependant, une toxicité variable.
Rechute
L'ibrutinib ou l'idelalisib peuvent être employés. La greffe de moelle peut être également proposé chez le sujet jeune.
Notes et références
- Triadou P, Quelques éléments d'histoire de l'hématologie biologique Annales de biologie clinique Vol 58;1: 19-28,Jan-fév 2000
- Rozman C, Montserrat E, Chronic lymphocytic leukemia. N Engl J Med, 1995;333:1052–105
- Hallek M, Shanafelt TD, Eichhorst B, Chronic lymphocytic leukaemia, Lancet, 2018;391:1524–1537
- Zenz T, Benner A, Dohner H, Stilgenbauer S, Chronic lymphocytic leukemia and treatment resistance in cancer: the role of the p53 pathway, Cell Cycle, 2008;7:3810–3814
- (en) Payelle-Brogard B, Dumas G, Magnac C et al. « Abnormal levels of the alpha chain of the CD22 adhesion molecule may account for low CD22 surface expression in chronic lymphocytic leukemia » Leukemia, 2006;20:877-878 DOI 10.1046/j.1365-2141.2002.03759.x
- Dighiero G, Hamblin TJ, [371:1017-1029 Chronic lymphocytic leukaemia], Lancet, 2008;371:1017-1029
- Casey R (2006) Étude cas-témoin multicentrique des facteurs de risque des hémopathies lymphoïdes (Doctoral dissertation, Dijon) (résumé)
- National Cancer Institute. SEER cancer statistics review 1975–2001
- Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics 2022, CA Cancer J Clin, 2022;72:7-33
- Sellick GS, Catovsky D, Houlston RS, Familial chronic lymphocytic leukemia, Semin Oncol, 2006;33:195-201
- Slager, SL, Benavente, Y, Blair, A et al. Medical history, lifestyle, family history, and occupational risk factors for chronic lymphocytic leukemia/small lymphocytic lymphoma: the InterLymph Non-Hodgkin Lymphoma Subtypes Project, J Natl Cancer Inst Monogr. 2014;2014:41–51
- Hamblin TJ, Have we been wrong about ionizing radiation and chronic lymphocytic leukemia?, Leuk Res, 2008;32:523-525
- Landgren O, Rapkin JS, Caporaso NE et al. Respiratory tract infections and subsequent risk of chronic lymphocytic leukemia, Blood, 2007;109:2198–2201
- https://www.sciencedirect.com/science/article/pii/S0013935105802439
- (en) Matutes E, Owusu-Ankomah K, Morilla R, Garcia Marco J, Catovsky D et al., « The immunological profile of B-cell disorders and proposal of a scoring system for the diagnosis of CLL », Leukemia, vol. 8, no 10, , p. 1640-5. (PMID 7523797)
- Rawstron AC, Bennett FL, O'Connor SJ et al. Monoclonal B-cell lymphocytosis and chronic lymphocytic leukemia, N Engl J Med, 2008;359:575–583
- Marti GE, Rawstron AC, Ghia P et al. Diagnostic criteria for monoclonal B-cell lymphocytosis, Br J Haematol, 2005;130:325–332
- (en) Marti GE, Rawstron AC, Ghia P, et al. Diagnostic criteria for monoclonal B-cell lymphocytosis, Br J Haematol, 2005;130:325-32 DOI 10.1111/j.1365-2141.2005.05550.x
- Clinical Staging of Chronic Lymphocytic Leukemia Rai KR, all Bloodjournal 1975 Aug;46(2):219-34
- A clinical staging system for chronic lymphocytic leukemia: prognostic significance Binet JL et col "Cancer" 1977 Aug;40(2):855-64.
- Hallek M, Langenmayer I, Nerl C et al. Elevated serum thymidine kinase levels identify a subgroup at high risk of disease-progression in early, non-smoldering chronic lymphocytic leukemia, Blood, 1999;93:1732–1737
- Hallek M, Wanders L, Ostwald M et al. Serum beta(2)-microglobulin and serum thymidine kinase are independent predictors of progression-free survival in chronic lymphocytic leukemia and immunocytoma, Leuk Lymphoma, 1996;22:439–447
- Zenz T, Eichhorst B, Busch R et al. TP53 mutation and survival in chronic lymphocytic leukemia, J Clin Oncol, 2010;28:4473–4479
- Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK, Unmutated Ig V-H genes are associated with a more aggressive form of chronic lymphocytic leukemia, Blood, 1999;94:1848–1854
- Rassenti LZ, Jain S, Keating MJ et al. Relative value of ZAP-70, CD38, and immunoglobulin mutation status in predicting aggressive disease in chronic lymphocytic leukemia, Blood, 2008;112:1923–1930
- Bulian P, Shanafelt TD, Fegan C et al. CD49d is the strongest flow cytometry-based predictor of overall survival in chronic lymphocytic leukemia, J Clin Oncol, 2014;32:897–904
- The International CLL IPI working group, An international prognostic index for patients with chronic lymphocytic leukaemia (CLL-IPI): a meta-analysis of individual patient data, Lancet Oncol, 2016;17:779–790
- Hallek M, Cheson BD, Catovsky D et al. iwCLL Guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL, Blood, 2018;131:2745-2760
- Burger J, Treatment of chronic lymphocytic leukemia, N Engl J Med, 2020;383:460-473
- Hallek M, Fischer K, Fingerle-Rowson G et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial, Lancet, 2010;376:1164–1174
- Goede V, Fischer K, Busch R et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions, N Engl J Med, 2014;370:1101–1110
- Byrd JC, Furman RR, Coutre SE et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia, N Engl J Med, 2013;369:32-42
- Ghia P, Pluta A, Wach M, et al. ASCEND: Phase III, randomized trial of acalabrutinib versus idelalisib plus rituximab or bendamustine plus rituximab in relapsed or refractory chronic lymphocytic leukemia, J Clin Oncol 2020;38:8015-8015
- « Idelalisib (Zydelig®) : restrictions concernant son utilisation dans le traitement de la leucémie lymphoïde chronique (LLC) et du lymphome folliculaire (LF) en rechute à la suite de nouveaux résultats d’essais cliniques- Lettre aux professionnels de santé - ANSM : Agence nationale de sécurité du médicament et des produits de santé », sur ansm.sante.fr (consulté le )
- Flinn IW, Hillmen P, Montillo M et al. The phase 3 DUO trial: duvelisib vs ofatumumab in relapsed and refractory CLL/SLL, Blood 2018;132:2446-2455
- Roberts AW, Davids MS, Pagel JM et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia, N Engl J Med, 2016;374:311–322
- Gauthier J, Hirayama AV, Purushe J et al. Feasibility and efficacy of CD19-targeted CAR T cells with concurrent ibrutinib for CLL after ibrutinib failure, Blood, 2020;135:1650-1660
- Eichhorst B, Fink AM, Bahlo J et al. First-line chemoimmunotherapy with bendamustine and rituximab versus fludarabine, cyclophosphamide, and rituximab in patients with advanced chronic lymphocytic leukaemia (CLL10): an international, open-label, randomised, phase 3, non-inferiority trial, Lancet Oncol, 2016;17:928–942
- Schweighofer CD, Ritgen M, Eichhorst BF et al. Consolidation with alemtuzumab improves progression-free survival in patients with chronic lymphocytic leukaemia (CLL) in first remission: long-term follow-up of a randomized phase III trial of the German CLL Study Group (GCLLSG), Br J Haematol, 2009;144:95–98
- Strati P, Lanasa M, Call TG et al. Ofatumumab monotherapy as a consolidation strategy in patients with previously untreated chronic lymphocytic leukaemia: a phase 2 trial, Lancet Haematol, 2016;3:e407–e414
- Greil R, Obrtlikova P, Smolej L et al. Rituximab maintenance versus observation alone in patients with chronic lymphocytic leukaemia who respond to first-line or second-line rituximab-containing chemoimmunotherapy: final results of the AGMT CLL-8a Mabtenance randomised trial, Lancet Haematol, 2016;3:e317–e329