Interaction non covalente
Une interaction non covalente diffère d'une liaison covalente en ce qu'elle n'implique pas le partage d'électrons, mais implique plutôt des variations plus dispersées des interactions électromagnétiques entre molécules ou au sein d'une molécule. L' énergie chimique libérée lors de la formation d'interactions non covalentes est généralement de l'ordre de 1 à 5 kcal / mol (1 000 à 5 000 calories pour 6,02 × 1023 molécules)[1]. Les interactions non covalentes peuvent être classées en différentes catégories, telles que les effets électrostatiques, les effets π, les forces de van der Waals et les effets hydrophobes[2].
Les interactions non covalentes sont essentielles pour maintenir la structure tridimensionnelle de grosses molécules, telles que les protéines et les acides nucléiques . De plus, elles sont également impliqués dans de nombreux processus biologiques dans lesquels de grosses molécules se lient spécifiquement entre elles (voir la section propriétés de la page ADN ). Ces interactions influencent également fortement la conception de médicaments, la cristallinité et la conception des matériaux, en particulier pour l' auto-assemblage et, en général, la synthèse de nombreuses molécules organiques[2] - [3] - [4] - [5].
Les forces intermoléculaires sont des interactions non covalentes qui se produisent entre différentes molécules, plutôt qu'entre différents atomes de la même molécule[2].
Interactions électrostatiques
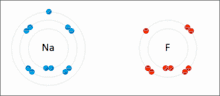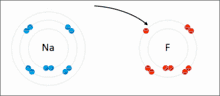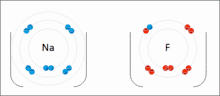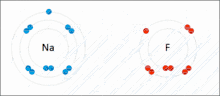
Les interactions ioniques impliquent l'attraction d'ions ou de molécules avec des charges permanentes de signes opposés. Par exemple, le fluorure de sodium implique l'attraction de la charge positive sur le sodium (Na+) avec la charge négative sur le fluorure (F−)[6]. Cependant, cette interaction particulière est facilement rompue lors de la solvatation dans l'eau ou d'autres solvants hautement polaires . Dans l'eau, l'appariement des ions est principalement entraîné par l'entropie; un seul pont de sel équivaut généralement à une valeur d'attraction d'environ ΔG = 5 kJ/mol à une force ionique intermédiaire I, à I proche de zéro la valeur augmente jusqu'à environ 8 kJ/mol. Les valeurs ΔG sont généralement additives et largement indépendantes de la nature des ions participants, à l'exception des ions de métaux de transition, etc. [7]
Ces interactions peuvent également être observées dans des molécules avec une charge localisée sur un atome particulier. Par exemple, la pleine charge négative associée à l'éthoxyde, la base conjuguée de l'éthanol, est le plus souvent accompagnée de la charge positive d'un sel de métal alcalin tel que le cation sodium (Na+).
Liaison hydrogène
Une liaison hydrogène est un type spécifique d'interaction qui implique une attraction dipôle-dipôle entre un atome d'hydrogène partiellement positif et un atome d'oxygène, d'azote, de soufre ou de fluor hautement électronégatif, partiellement négatif (non lié de manière covalente audit hydrogène atome). Il ne s'agit pas d'une liaison covalente, mais plutôt d'une forte interaction non covalente. Elle explique pourquoi l'eau est un liquide à température ambiante et non un gaz (étant donné le faible poids moléculaire de l' eau). Le plus souvent, la force des liaisons hydrogène se situe entre 0 et 4 kcal/mol, mais peut parfois atteindre 40 kcal/mol[2]. Dans les solvants tels que le chloroforme ou le tétrachlorure de carbone, on observe par exemple pour l’interaction entre les amides des valeurs additives environ 5 kJ/mol. Selon Linus Pauling, la force d'une liaison hydrogène est essentiellement déterminée par les charges électrostatiques. Les mesures de milliers de complexes dans le chloroforme ou le tétrachlorure de carbone ont conduit à des incréments d'énergie sans additif pour tous les types de combinaisons donneur-accepteur[8] - [9].
Liaison halogène
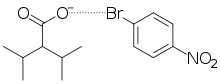
La liaison halogène est un type d'interaction non covalente qui n'implique ni la formation ni la rupture de liaisons réelles, mais est plutôt similaire à l'interaction dipôle-dipôle connue sous le nom de liaison hydrogène . Dans la liaison halogène, un atome d' halogène agit comme une espèce électrophile ou chercheuse d'électrons et forme une faible interaction électrostatique avec une espèce nucléophile ou riche en électrons. L'agent nucléophile dans ces interactions a tendance à être hautement électronégatif (tel que l'oxygène, l'azote ou le soufre), ou peut être anionique, portant une charge formelle négative. Par rapport à la liaison hydrogène, l'atome d'halogène prend la place de l'hydrogène partiellement chargé positivement en tant qu'électrophile.
La liaison halogène ne doit pas être confondue avec les interactions halogène-aromatique, car les deux diffèrent par définition. Les interactions halogène-aromatique impliquent un nuage π aromatique riche en électrons en tant que nucléophile; la liaison halogène est limitée aux nucléophiles monoatomiques[3].
Forces de Van der Waals
Les forces de Van der Waals sont un sous-ensemble des interactions électrostatiques impliquant des dipôles permanents ou induits (ou multipôles). Ceux-ci comprennent les éléments suivants:
- interactions dipôle – dipôle permanentes, appelées aussi force de Keesom
- interactions dipolaires induites par les dipôles, ou force de Debye
- les interactions dipolaires induites par les dipôles, communément appelées forces de dispersion de London
La liaison hydrogène et la liaison halogène ne sont généralement pas classées comme des forces de Van der Waals
Dipôle – dipôle
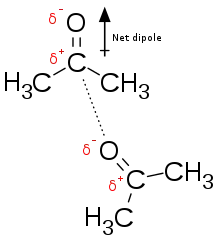
Les interactions dipôle – dipôle sont des interactions électrostatiques entre les dipôles permanents des molécules. Ces interactions ont tendance à aligner les molécules pour augmenter leur attraction (réduisant l'énergie potentielle). Normalement, les dipôles sont associés à des atomes électronégatifs, notamment l' oxygène, l' azote, le soufre et le fluor .
Par exemple, l'acétone, l'ingrédient actif de certains dissolvants pour vernis à ongles, possède un dipôle net associé au carbonyle (voir figure 2). Puisque l'oxygène est plus électronégatif que le carbone qui lui est lié de manière covalente, les électrons associés à cette liaison seront plus proches de l'oxygène que du carbone, créant une charge négative partielle (δ−) sur l'oxygène et une charge positive partielle (δ+) sur le carbone. Ce ne sont pas des charges complètes car les électrons sont toujours partagés par une liaison covalente entre l'oxygène et le carbone. Si les électrons n'étaient plus partagés, alors la liaison oxygène-carbone serait une interaction électrostatique.
Les molécules contiennent souvent des groupes dipolaires, mais n'ont pas de moment dipolaire global. Cela se produit s'il y a une symétrie dans la molécule qui provoque l'annulation des dipôles. Cela se produit dans des molécules telles que le tétrachlorométhane. Notez que l'interaction dipôle-dipôle entre deux atomes individuels est généralement nulle, car les atomes portent rarement un dipôle permanent.
Dipôle induit par dipôle
Une interaction dipolaire induite par un dipôle (force de Debye) est due à l'approche d'une molécule avec un dipôle permanent vers une autre molécule non polaire sans dipôle permanent. Cette approche amène les électrons de la molécule non polaire à être polarisés vers ou à l'écart du dipôle (ou "induire" un dipôle) de la molécule qui s'approche[10]. Plus précisément, le dipôle peut provoquer une attraction électrostatique ou une répulsion des électrons de la molécule non polaire, en fonction de l'orientation du dipôle entrant. Les atomes avec des rayons atomiques plus grands sont considérés comme plus «polarisables» et subissent donc de plus grandes attractions en raison de la force de Debye.
Forces de dispersion de London
Les forces de dispersion de London [11] - [12] - [13] - [14] sont le type d'interaction non covalente le plus faible. Dans les molécules organiques, cependant, la multitude de contacts peut conduire à des contributions plus importantes, notamment en présence d'hétéroatomes. Elles sont également connues sous le nom d'"interactions dipolaires induites par les dipôles induits" et présentes entre toutes les molécules, même celles qui, par nature, n'ont pas de dipôles permanents. Les interactions dispersives augmentent avec la polarisabilité des groupes en interaction, mais sont affaiblies par des solvants de polarisabilité accrue[15]. Ils sont causés par la répulsion temporaire des électrons loin des électrons d'une molécule voisine, conduisant à un dipôle partiellement positif sur une molécule et un dipôle partiellement négatif sur une autre molécule[4]. L'hexane est un bon exemple de molécule sans polarité ou avec des atomes hautement électronégatifs, mais c'est un liquide à température ambiante principalement en raison des forces de dispersion de London. Dans cet exemple, lorsqu'une molécule d'hexane s'approche d'une autre, un dipôle partiellement négatif faible temporaire sur l'hexane peut polariser le nuage d'électrons d'un autre hexane, provoquant un dipôle partiellement positif sur cette molécule d'hexane. En l'absence de solvants, les hydrocarbures tels que l'hexane forment des cristaux en raison des forces de dispersion; la chaleur de sublimation des cristaux est une mesure de l'interaction dispersive. Bien que ces interactions soient de courte durée et très faibles, elles peuvent expliquer pourquoi certaines molécules non polaires sont des liquides à température ambiante.
π-effets
Les effets π peuvent être décomposés en de nombreuses catégories, y compris les interactions π-π, les interactions cation-π, les interactions anion-π et les interactions polaires-π. En général, les effets π sont associés aux interactions de molécules avec les systèmes π de molécules conjuguées comme le benzène[2].
Interaction π – π
Les interactions π – π sont associées à l'interaction entre les orbitales π d'un système moléculaire[2]. La haute polarisabilité des cycles aromatiques conduit à des interactions dispersives comme contribution majeure aux effets dits d'empilement . Ceux-ci jouent un rôle majeur pour les interactions des nucléobases, par exemple dans l'ADN[16]. Pour un exemple simple, un cycle benzénique, avec son nuage π entièrement conjugué, interagira de deux manières principales (et une manière mineure) avec un cycle benzénique voisin par une interaction π – π (voir figure 3). Les deux principales façons dont le benzène s'empile sont bord à face, avec une enthalpie de ~ 2 kcal/mol, et déplacées (ou empilées en barbotine), avec une enthalpie de ~ 2,3 kcal/mol. La configuration sandwich n'est pas aussi stable que les deux interactions mentionnées précédemment en raison de la forte répulsion électrostatique des électrons dans les orbitales π.
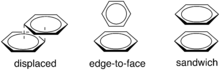
Interaction cation – π et anion – π
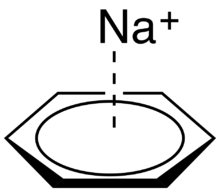
Les interactions cation – pi impliquent la charge positive d'un cation interagissant avec les électrons dans un système π d'une molécule[2]. Cette interaction est étonnamment forte (aussi forte ou plus forte que la liaison hydrogène dans certains contextes), et a de nombreuses applications potentielles dans les capteurs chimiques[17]. Par exemple, l'ion sodium peut facilement s'asseoir au sommet du nuage π d'une molécule de benzène, avec une symétrie C 6 (voir figure 4).
Les interactions anion – π sont similaires aux interactions cation – π, mais inversées. Dans ce cas, un anion se trouve au sommet d'un système π pauvre en électrons, généralement établi par le placement de substituants attracteurs d'électrons sur la molécule conjuguée[18].
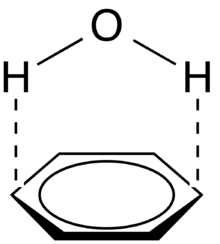
Polaire – π
Les interactions polaires – π impliquent des molécules avec des dipôles permanents (comme l'eau) interagissant avec le moment quadripolaire d'un système π (comme celui du benzène, voir figure 5). Bien qu'elles ne soient pas aussi fortes qu'une interaction cation-π, ces interactions peuvent être assez fortes (~ 1-2 kcal/mol) et sont couramment impliquées dans le repliement des protéines et la cristallinité des solides contenant à la fois des liaisons hydrogène et des systèmes π[2]. En fait, toute molécule avec un donneur de liaison hydrogène (hydrogène lié à un atome hautement électronégatif) aura des interactions électrostatiques favorables avec le système π riche en électrons d'une molécule conjuguée.
Effet hydrophobe
L' effet hydrophobe est le désir de molécules non polaires de s'agréger dans des solutions aqueuses afin de se séparer de l'eau[19]. Ce phénomène conduit à une surface minimale des molécules non polaires exposée aux molécules d'eau polaires (généralement des gouttelettes sphériques), et est couramment utilisé en biochimie pour étudier le repliement des protéines et divers autres phénomènes biologiques. L'effet est également généralement observé lors du mélange de diverses huiles et de l'eau. Au fil du temps, l'huile reposant au-dessus de l'eau commencera à s'agréger en grandes sphères aplaties à partir de gouttelettes plus petites, conduisant finalement à un film de toute l'huile reposant sur une piscine d'eau. Cependant l'effet hydrophobe n'est pas considéré comme une interaction non covalente car il est fonction de l'entropie et non d'une interaction spécifique entre deux molécules, généralement caractérisée par une compensation entropy/enthalpie[20] - [21] - [22]. Un effet hydrophobe essentiellement enthalpique se matérialise si un nombre limité de molécules d'eau est restreint dans une cavité; le déplacement de telles molécules d'eau par un ligand libère ces molécules d'eau qui bénéficient alors dans la masse d'eau d'un maximum de liaisons hydrogène voisines de quatre[23] - [24].
Exemples
Conception de médicaments
La plupart des médicaments pharmaceutiques sont de petites molécules qui provoquent une réponse physiologique en "se liant" à des enzymes ou à des récepteurs, provoquant une augmentation ou une diminution de la capacité de l'enzyme à fonctionner. La liaison d'une petite molécule à une protéine est régie par une combinaison de considérations stériques ou spatiales, en plus de diverses interactions non covalentes, bien que certains médicaments modifient de manière covalente un site actif (voir inhibiteurs irréversibles ). En utilisant le «modèle de verrou et clé» de la liaison enzymatique, un médicament (clé) doit avoir à peu près les dimensions appropriées pour s'adapter au site de liaison de l'enzyme (verrou)[25]. En utilisant le modèle moléculaire de taille appropriée, les médicaments doivent également interagir avec l'enzyme de manière non covalente afin de maximiser la constante de liaison d'affinité de liaison et de réduire la capacité du médicament à se dissocier du site de liaison. Ceci est réalisé en formant diverses interactions non covalentes entre la petite molécule et les acides aminés dans le site de liaison, notamment : liaison hydrogène, interactions électrostatiques, empilement pi, interactions de van der Waals et interactions dipôle – dipôle.
Des médicaments métalliques non covalents ont été développés. Par exemple, des composés tri-hélicoïdaux dinucléaires dans lesquels trois brins de ligand s'enroulent autour de deux métaux, résultant en une tétracation à peu près cylindrique, ont été préparés. Ces composés se lient aux structures d'acide nucléique moins courantes, telles que l'ADN duplex, les structures de fourche en forme de Y et les jonctions à 4 voies[26].
Pliage et structure des protéines
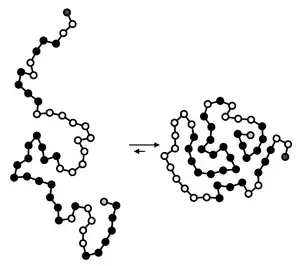
Le repliement de la plupart des protéines d'une séquence primaire (linéaire) d'acides aminés à une structure tridimensionnelle est régi par de nombreux facteurs, y compris des interactions non-covalentes. Les ~ 5 premières millisecondes de repliement dépendent principalement des forces de van der Waals, par quoi la protéine se replie de manière à orienter les acides aminés non polaires à l'intérieur de la protéine globulaire, tandis que les résidus d'acides aminés plus polaires sont exposés à un solvant aqueux. Cette phase est connue sous le nom d' effondrement hydrophobe, lorsque les interactions non polaires non covalentes excluent l'eau de l'intérieur de la structure protéique 3D en développement.
Après cette « phase de salve » initiale, des interactions non covalentes plus polaires prennent le relais. Entre 5 et 1 000 millisecondes après le début du repliement des protéines, les structures tridimensionnelles des protéines, appelées structures secondaires et tertiaires, sont stabilisées par la formation de liaisons hydrogène, en plus de ponts disulfure (liaisons covalentes). Grâce à une série de petits changements conformationnels, les orientations spatiales sont modifiées de manière à arriver à l'orientation la plus énergétiquement minimisée possible. Le repliement des protéines est souvent facilité par des enzymes appelées chaperons moléculaires[27]. Les stériques, la contrainte de liaison et la contrainte angulaire jouent également un rôle majeur dans le repliement d'une protéine de sa séquence primaire à sa structure tertiaire.
Des structures protéiques tertiaires uniques peuvent également s'assembler pour former des complexes protéiques composés de plusieurs sous-unités repliées indépendamment. Dans son ensemble, cela s'appelle la structure quaternaire d'une protéine. La structure quaternaire est générée par la formation d'interactions non covalentes relativement fortes, telles que des liaisons hydrogène, entre différentes sous-unités pour générer une enzyme polymère fonctionnelle[28]. Certaines protéines utilisent également des interactions non covalentes pour lier des cofacteurs dans le site actif pendant la catalyse, mais un cofacteur peut également être lié de manière covalente à une enzyme. Les cofacteurs peuvent être des molécules organiques ou inorganiques qui contribuent au mécanisme catalytique de l'enzyme active. La force avec laquelle un cofacteur est lié à une enzyme peut varier considérablement ; Les cofacteurs liés de manière non covalente sont généralement ancrés par des liaisons hydrogène ou des interactions électrostatiques .
Points d'ébullition
Les interactions non covalentes ont un effet significatif sur le point d'ébullition d'un liquide. Le point d'ébullition est défini comme la température à laquelle la pression de vapeur d'un liquide est égale à la pression entourant le liquide. Plus simplement, c'est la température à laquelle un liquide devient un gaz. Comme on pouvait s'y attendre, plus les interactions non covalentes présentes pour une substance sont fortes, plus son point d'ébullition est élevé. Par exemple, considérons trois composés de composition chimique similaire: le n-butoxyde de sodium (C 4 H 9 ONa), l'éther diéthylique (C 4 H 10 O) et le n-butanol (C 4 H 9 OH).
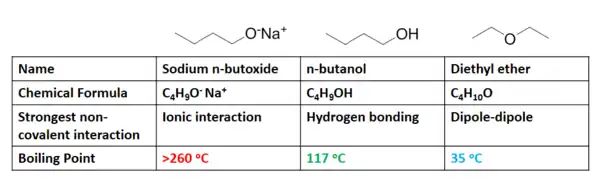
Les interactions non covalentes prédominantes associées à chaque espèce en solution sont répertoriées dans la figure ci-dessus. Comme discuté précédemment, les interactions ioniques nécessitent beaucoup plus d'énergie pour se rompre que les liaisons hydrogène, qui à leur tour nécessitent plus d'énergie que les interactions dipôle – dipôle. Les tendances observées dans leurs points d'ébullition (figure 8) montrent exactement la corrélation attendue, où le n-butoxyde de sodium nécessite beaucoup plus d'énergie thermique (température plus élevée) pour bouillir que le n-butanol, qui bout à une température beaucoup plus élevée que l'éther diéthylique. L'énergie thermique requise pour qu'un composé passe du liquide au gaz est associée à l'énergie nécessaire pour briser les forces intermoléculaires que chaque molécule subit dans son état liquide.
Références
- Noncovalent bonds – Molecular Cell Biology (textbook), Lodish, Berk, Zipursky, Matsudaira, Baltimore, Darnell.
- Eric Anslyn, Modern Physical Organic Chemistry, Sausalito, CA, University Science, (ISBN 978-1-891389-31-3)
- Cockroft et Hunter, Christopher A., « Chemical double-mutant cycles: dissecting non-covalent interactions », Chemical Society Reviews, vol. 36, no 2, , p. 172–88 (PMID 17264921, DOI 10.1039/b603842p)
- Theodore Brown, Chemistry : the central science, Upper Saddle River, NJ, 11th, (ISBN 978-0-13-600617-6)
- (en) Matthew Eisler, Encyclopedia of nanoscience and society, Thousand Oaks, Calif., Sage, (ISBN 978-1-4129-7209-3, DOI 10.4135/9781412972093.n412), « Self-Assembly »
- Ionic Interactions in Natural and Synthetic Macromolecules, John Wiley & Sons, (ISBN 978-0-470-52927-0)
- Schneider, « Binding mechanisms in supramolecular complexes », Angew. Chem. Int. Ed. Engl., vol. 48, no 22, , p. 3924–3977 (PMID 19415701, DOI 10.1002/anie.200802947)
- Abraham, « Scales of Solute Hydrogen-bonding: their Construction and Application to Physicochemical and Biochemical Processes », Chem. Soc. Rev., vol. 22, no 2, , p. 73–83 (DOI 10.1039/CS9932200073)
- Raevsky et Skvortsov, « Quantifying Hydrogen Bonding », J. SAR and QSAR Environment. Res., vol. 16, no 3, , p. 287–300 (PMID 15804815, DOI 10.1080/10659360500036893)
- « Induced-Dipole Forces » (consulté le )
- Noncovalent Forces, Springer, (ISBN 978-3-319-14162-6)
- Non-covalent Interactions in Quantum Chemistry and Physics: Theory and Applications A.Otero de la Roza, G. A. DiLabio, (Eds), Elsevier; 2017, (ISBN 012809835X)
- Non-covalent Interactions in the Synthesis and Design of New Compounds A M. Maharramov, K. T. Mahmudov, M. N. Kopylovich, A. J. L. Pombeiro Wiley; 2016, (ISBN 9781119109891)
- P. Hobza and K. Müller-Dethlefs Non-Covalent Interactions: Theory and Experiment (Theoretical and Computational Chemistry Series) Royal Society of Chemistry; 2009, (ISBN 1847558534)
- Schneider, « Dispersive Interactions in Solution Complexes Dispersive Interactions in Solution Complexes », Acc. Chem. Res, vol. 48, no 7, , p. 1815–1822 (PMID 26083908, DOI 10.1021/acs.accounts.5b00111)
- Riley et Hobza, « On the Importance and Origin of Aromatic Interactions in Chemistry and Biodisciplines », Acc. Chem. Res., vol. 46, no 4, , p. 927–936 (PMID 22872015, DOI 10.1021/ar300083h)
- Sastry et Mahadevi, « Cation-π interaction: Its role and relevevance in chemistry, biology, and material science », Chemical Reviews, vol. 113, , p. 2100 (PMID 23145968, DOI 10.1021/cr300222d)
- Quiñonero, Garau, Carolina, Rotger, Carmen et Frontera, Antonio, « Anion–π Interactions: Do They Exist? », Angewandte Chemie International Edition, vol. 41, no 18, , p. 3389–3392 (PMID 12298041, DOI 10.1002/1521-3773(20020916)41:18<3389::AID-ANIE3389>3.0.CO;2-S)
- IUPAC, Compendium of Chemical Terminology, (ISBN 978-0-9678550-9-7, DOI 10.1351/goldbook.H02907), « Hydrophobic interaction »
- Kronberg, « The hydrophobic effect », Curr. Opinion Coll. Interface Sci., vol. 22, , p. 14–22 (DOI 10.1016/j.cocis.2016.02.001)
- Hillyer et Gibb, « Molecular Shape and the Hydrophobic Effect », Annu. Rev. Phys. Chem., vol. 67, , p. 307–329 (PMID 27215816, PMCID 5571648, DOI 10.1146/annurev-physchem-040215-112316)
- Ben-Amotz, « Water-Mediated Hydrophobic Interactions », Annu. Rev. Phys. Chem., vol. 67, , p. 617–638 (PMID 27215821, DOI 10.1146/annurev-physchem-040215-112412)
- Snyder, Lockett, Moustakas et Whitesides, « Is it the shape of the cavity, or the shape of the water in the cavity? », Eur. Physical J. Spec. Topics, vol. 223, no 5, , p. 853–891 (DOI 10.1140/epjst/e2013-01818-y, lire en ligne)
- Biedermann, Nau et Schneider, « The Hydrophobic Effect Revisited—Studies with Supramolecular Complexes Imply High‐Energy Water as a Noncovalent Driving Force », Angew. Chem. Int. Ed. Engl., vol. 53, no 42, , p. 2–16 (PMID 25070083, DOI 10.1002/anie.201310958)
- « Biomolecules: Enzymes », ChemPages Netorials, University of Wisconsin - Madison (consulté le )
- Lucia Cardo et Michael J. Hannon, Metallo-Drugs: Development and Action of Anticancer Agents, vol. 18, Berlin, de Gruyter GmbH, , 303–324 p. (ISBN 9783110470734, PMID 29394030, DOI 10.1515/9783110470734-017), « Chapter 11. Non-covalent Metallo-Drugs: Using Shape to Target DNA and RNA Junctions and Other Nucleic Acid Structures »
- Donald., & Voet, Judith G. Voet, Biochemistry, Hoboken, NJ, 4th, (ISBN 978-0-470-57095-1)
- Richard B. Silverman, The organic chemistry of drug design and drug action, Amsterdam [u.a.], 2., (ISBN 978-0-12-643732-4, lire en ligne)