Solvatation
La solvatation est le phénomène physico-chimique observé lors de la dissolution d'une substance chimique dans un solvant.
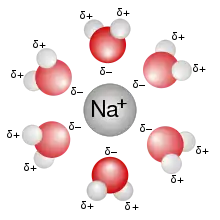
Lors de l'introduction d'une espèce chimique initialement à l'état solide (sous forme de cristal ou amorphe), liquide ou gazeux dans un solvant, les atomes, ions ou molécules de l'espèce chimique se dispersent dans la solution et interagissent avec les molécules de solvant. Cette interaction s'appelle la solvatation. Elle est de différente nature suivant le soluté et le solvant et recouvre des phénomènes aussi différents que des interactions ion-dipôle (soluté : Na+, solvant : eau), des liaisons hydrogène (soluté alcool, solvant eau) ou des liaisons de van der Waals (soluté méthane, solvant cyclohexane).
Quand le soluté est en phase condensée (liquide ou solide), la solvatation entre dans le bilan énergétique qui met en jeu la séparation des molécules ou des ions dans le soluté avant sa dispersion dans le solvant. Le soluté ne se dissout que si les interactions soluté-solvant compensent la perte des interactions soluté-soluté et solvant-solvant du fait de la dissolution :
- soit par réaction chimique ;
- soit en affaiblissant suffisamment les liaisons (par exemple, l'eau divise les forces électrostatiques par environ 80).
Pour que la dissolution ait lieu, il faut que les molécules du solvant aient suffisamment d'affinité avec celles du soluté.
Thermodynamique de la dissolution
Points de vue microscopique et macroscopique
D'un point de vue microscopique, si les molécules du solvant entourent le soluté, c'est que les interactions solvant-soluté sont suffisamment stables. Cette organisation de la molécule de soluté entourée de molécules de solvant participe à la structuration de la solution.
Du point de vue macroscopique, la dissolution met en jeu deux types d'énergie :
- une enthalpie de dissolution, bilan des liaisons rompues (soluté-soluté et solvant-solvant) et des liaisons formées (soluté-solvant) ;
- une entropie de dissolution, résultant du désordre dû à la dispersion du soluté dans le solvant, et de l'ordre créé avec la structuration du solvant (c'est-à-dire la solvatation).
La composante enthalpique se traduit par la chaleur de dissolution, et le bilan enthalpique plus entropique se traduit par l'enthalpie libre de dissolution, c'est-à-dire la solubilité, donc la quantité plus ou moins grande de soluté qui peut être dissous.
Énergies mises en jeu lors de la dissolution
Examinons le cas où le soluté est solide ionique avant la dissolution.
- L'énergie de cohésion du solide : on doit fournir de l'énergie pour vaincre cette cohésion. Dans le cas d'un solide ionique comme un sel alcalin, par exemple NaCl, l'enthalpie de sublimation du solide mise en jeu est −Er, où Er est l'énergie réticulaire du solide.
- L'énergie de solvatation : elle est présente lors de l'attraction entre les entités dissoutes, porteuses soit de charges ioniques ou de dipôles induits, et les molécules plus ou moins polaires du solvant. La solvatation est toujours exothermique : son enthalpie est donc toujours négative. Elle est de grandeur comparable à l'énergie de cohésion de cristaux ioniques et peut ainsi permettre leur dissolution.
- L'énergie de changement de structure du solvant : pour que les molécules de solvant puissent s'attacher aux entités dissoutes dans le processus de solvatation, et pour que les molécules de soluté puissent même s'insérer dans la structure « molle » qui prévalait dans le solvant, il faut dissocier cette structure, aussi non rigide soit-elle. Ceci demande une certaine énergie, apportée le plus souvent par hausse de la température. C'est la grandeur de cette énergie qui régit surtout la solubilité des solides moléculaires. Elle limite la solubilité de composés covalents non polaires.
- On peut noter que le chauffage dans un environnement soumis à la pesanteur apporte aussi des mouvements de convection dans le soluté lors des échanges de chaleur (ainsi que l'éventuelle évaporation partielle du solvant, dont les bulles de gaz, plus volumineuses que le solvant liquide, propulsent des quantités importantes de la phase liquide non évaporée ; cette intensité des mouvements de convection augmente aussi lorsque les bulles gazeuses sont formées et qu'elles remontent à la surface du liquide, en faisant descendre le solvant vers le fond ; cet effet d'accélération de la convection est réduit si le chauffage se fait par le côté ou par le haut, car le gaz se forme directement à la surface du solvant qui n'est alors soumis qu'à la convection du liquide lui-même ; toutefois le chauffage induit aussi des dilatations du liquide dont la masse volumique diminue aussi et, là encore, produit des mouvements de convection réduisant les écarts de température). En l'absence de pesanteur, seule la dilatation apporte des mouvements mécaniques (uniformes), les échanges de température dépendant alors de la viscosité du liquide (limitant la vitesse de propagation du changement de température) et de son agitation aléatoire propre (augmentée par la température qui réduit aussi la viscosité). Tous ces mouvements favorisent aussi la dispersion du soluté et des produits de solvatation et la vitesse de solvatation.
- Dans le cas de composés polaires, les forces de cohésion dans le solide et les interactions soluté/solvant (énergie de solvatation) sont plus grandes. Cette énergie, liée au changement de structure du solvant, devient moins importante, et la solubilité augmente. L'énergie de changement de structure du solvant est négligeable dans le cas des solides ioniques, étant complètement dominée par les deux premiers types d'énergie.
La solvatation d'une espèce dépend de la nature du solvant et du soluté : en règle générale, un composé polaire sera très bien solvaté dans un solvant polaire, tandis qu'un composé apolaire sera mieux solvaté dans un solvant apolaire.
La solvatation dans l'eau est appelée « hydratation ».
Structure de la solution
Couches de solvatation
L'ion (ou la molécule) solvaté est entouré de molécules de solvant qui lui sont directement liées, qui sont orientées (dans le cas d'une interaction ion-dipôle), et qui ne s'échangent que lentement avec des molécules extérieures à cette première sphère de solvatation.
Cette première sphère est entourée d'autres molécules de solvant qui sont non directement liées à l'ion, qui sont peu orientées, qui s'échangent rapidement avec des molécules de solvant du reste de la solution, et qui s'échangent lentement avec les molécules de la première sphère.
Le reste de la solution est constitué de molécules de solvant qui sont dans le même état qu'en l'absence de soluté. La littérature anglo-saxonne appelle ces molécules le bulk.
Si cette structuration de la solution est commune aux différents solvants et solutés, le nombre de molécules de solvant dans chaque sphère de solvatation est propre à chaque type de solution.
Nombre de solvatation
Le nombre de solvatation est le nombre (sans unité) de molécules de la première sphère de solvatation ou, suivant les cas, le nombre total de molécules de solvant des deux couches de solvatation. Le nombre de solvatation dans la première sphère est une notion assez facile à définir, étant donné la stabilité des interactions solvant-soluté. Dans le cas d'un aquacomplexe, ce nombre est la coordinence du cation central. C'est 6 pour un grand nombre d'ions, et en particulier pour tous les ions de transitions. Pour les petits alcalins, c'est moins, jusqu'à 4 pour l'ion lithium Li+.
Le nombre de solvatation total, incluant les molécules de solvant de la seconde sphère de solvatation, est plus délicat à définir. Il est le plus souvent défini par la méthode utilisée pour le mesurer[1]. Cette disparité, qui peut paraître perturbante en première instance, traduit en fait l'importance de la notion de nombre de solvatation dans des situations variées.
Les méthodes de détermination des nombres de solvatation sont aussi variées que :
- les méthodes de diffraction (rayons X, neutrons) ;
- les calculs théoriques ;
- les dilutions isotopiques ;
- la RMN ;
- les spectroscopies raman, UV-visible ;
- les mesures thermodynamiques ;
- etc.
Les nombres de solvatation par des molécules d'eau dans la première sphère sont (détermination par diffraction de rayons X ou de neutrons, les valeurs peuvent être dépendantes de la température) :
- 4 pour Li+, H3O+, Be2+
- 4 à 8 pour Na+, K+, Cs+
- 6 pour Mg2+, Ca2+, Mn2+, Fe2+, etc.
- 6 pour F−, Cl−, Br−
- 6 à 7 pour I−
- 6 à 9 pour NO3−
- 8 pour NH4+, Eu3+, Gd3+, Tb3+, etc.
- 8 pour SO42−
- 9 pour Pr3+, Sm3+
Surface de la solution
Si une molécule ou un ion de soluté se retrouve à la surface du liquide, il ne disposera pas de la totalité de son « bouclier » de molécules de solvant. D'un point de vue thermochimique, son énergie sera plus élevée : la molécule (ou l'ion) ne sera que partiellement solvatée. Par conséquent, l'extrême surface de la solution — les premières couches moléculaires — est constituée de solvant quasiment pur.
Mobilité des espèces
Une espèce solvatée est entourée par une couche de molécules de solvant. L'espèce solvatée est donc plus grosse que l'espèce seule, sa mobilité est donc plus faible.
Cette sphère de solvatation est d'autant plus grande que l'ion est petit. Ainsi, le volume efficace de l'ion Na+ dans l'eau (c'est-à-dire le volume de l'ion et de sa sphère de solvatation) est plus grand que celui de l'ion K+, qui a pourtant un rayon ionique plus grand.
Notes et références
- (en) Yizhak Marcus, Ion solvatation, éd. J. Wiley & Sons, 1985.