Monochlorure de xénon
Le monochlorure de xénon est le chlorure de xénon de formule XeCl, connu à l'état gazeux, mais instable.
| Monochlorure de xénon | ||
| ||
 | ||
| Identification | ||
|---|---|---|
| No CAS | ||
| SMILES | ||
| InChI | ||
| Propriétés chimiques | ||
| Formule | XeCl | |
| Masse molaire[1] | 166,746 ± 0,008 g/mol Cl 21,26 %, Xe 78,74 %, |
|
| Unités du SI et CNTP, sauf indication contraire. | ||
XeCl est une molécule exciplexe, synthétisée pour la première fois en 1965[2] à l'université d'État du Kansas. Son existence est supposée depuis le XXe siècle[3]. En effet, la configuration électronique du xénon excité est analogue à celle des métaux alcalins, éléments pouvant se lier avec des halogènes. Les phénomènes d'apparition et de disparition du monochlorure de xénon sont de l'ordre de quelques nanosecondes, ce qui en fait une molécule au schéma cinétique complexe.
La principale utilisation du monochlorure de xénon est médicale, via l'utilisation de lasers à excimère de longueur d'onde 308 nm.
Introduction
Les molécules homonucléaires n'étant stables que dans des états électroniquement excités sont appelées excimères. Ainsi, de manière analogue, les molécules hétéronucléaires n'étant stables que dans des états électroniquement excités tels que XeCl sont appelés exciplexes.
Le chlorure de xénon appartient à la classe des halogénures de gaz rares, notés RgX. Ces molécules se désexcitent en émettant un photon dont l’énergie est de quelques eV et dont la longueur d’onde se situe dans le visible ou l’ultraviolet.
Les gaz ou mélanges de gaz pouvant conduire à la formation d'halogénures de gaz rares constituent un milieu laser quasi-idéal puisque l’inversion de population est obtenue directement lors de la formation de l'exciplexe. L’autre conséquence de la nature instable des halogénures de gaz rares à l'état fondamental est que les espèces excimères ou exciplexes ne constituent pas elles-mêmes le milieu laser mais doivent être générées en temps réel par une excitation extérieure (décharge, faisceaux d’électrons, micro-ondes, particules alpha). Au moins deux gaz différents doivent être utilisés afin de générer des exciplexes : un gaz donneur d’halogène et un gaz rare. Les principaux gaz donneurs de chlore employés dans les lasers sont HCl et CCl4. Cl2 se forme dans le milieu laser.
Toutes les molécules d’halogénures de gaz rares ne conduisent pas forcément à des développements de lasers (voir Tableau 1). L'existence de certains d'entre eux est même impossible. Les halogénures de gaz rares et leurs applications ont fait l’objet de plusieurs mises au point bibliographiques[4] - [5] - [6] - [7] - [8] - [9] - [10] - [11] - [12] - [13]. Compte tenu des progrès récents, ces travaux anciens doivent être actualisés régulièrement.
Tableau 1 : Caractéristiques des molécules d’halogénures de gaz rares. Le radon n’est pas pris en compte.
- D indique que la molécule ne peut exister car elle est dissociative.
- F signale une fluorescence observée.
- L désigne une molécule dont l’effet laser est démontré[6].
Plusieurs articles de synthèse relatifs à la technologie des lasers à chlorure de xénon et à leurs applications ont été publiés[14] - [15] - [16] - [17] - [18] - [19]. Les halogénures de gaz rares ont des interactions complexes nécessitant une étude en profondeur de la cinétique du milieu laser lors de leur utilisation[14] - [19] - [20]. Des résultats récents ont permis de mieux comprendre la physico-chimie du milieu laser[21] - [22] - [23] - [24]. L’investigation spectroscopique des gaz RgX se limite à la zone visible-proche ultraviolet, qui est la longueur d'onde émettrice de ce type de gaz. Les molécules de chlorure de xénon qui présentent le plus d’intérêt pour les applications laser sont XeCl et Xe2Cl.
La faisabilité de sources de lumière incohérentes a récemment été démontrée à partir de décharges dans les mélanges xénon/donneur de chlore à basse pression. Ces lampes à décharge sont fiables, d’un fonctionnement simple, et d’un coût peu élevé. Cela en fait des concurrents directs des sources laser[25] - [26].
Histoire
L’idée que les gaz rares puissent contribuer à la formation d’halogénures remonte au début des années 1920 [3]. Andreas von Antropoff[27] puis Giuseppe Oddo (en)[28] ont indiqué la possibilité pour le krypton et le xénon de fournir des bromures et des chlorures. Yost et Kaye en 1933[29] ont été les premiers à tenter la synthèse du chlorure de xénon ; ils ont soumis un mélange de xénon (70 torr) et de chlore (225 torr), placé dans un tube en quartz, au flux lumineux d’une lampe à vapeur de mercure. Cette expérience s’est soldée par un échec (vraisemblablement en raison des très faibles quantités de XeCl formées).
La première synthèse de chlorure de xénon (XeCl et XeCl2) semble dater de 1965[30]. Pendant une dizaine d’années, ce sont les molécules XeCl2 et XeCl4 (formées à quelques kelvins donc à l’état solide) qui ont retenu l’attention des chercheurs. Récemment, Prosperio et al.[31] ont monté que l’existence de XeCl2 à l’état gazeux était possible et que cette molécule joue un rôle dans la cinétique du laser. Cependant XeCl2 ne semble émettre que dans l’infrarouge qui ne fait pas l’objet de l'investigation spectroscopique.
En 1973, Riveros et al.[32] ont réussi la synthèse de l’ion XeCl− en phase gazeuse pour une pression de 10-4 torr. Cet ion moléculaire n’a par la suite suscité que peu d’intérêt.
En 1975, Velazco et Setser[33] présentent un spectre comportant une émission située à 304 nm qu’ils attribuent à XeCl*. Cette émission a été obtenue en mélangeant des atomes de xénon Xe(3P2) avec du gaz chlore Cl2 ou avec d’autres composés chlorés (NOCl et SOCl2) ; l’excitation était assurée par une décharge à cathode froide et la pression totale était de quelques torr. Quelques mois plus tard Ewing et Brau[34] mettent en évidence l’effet laser sur une bande 2Σ1/2+ → 2Σ1/2+ de XeCl à 308 nm. Le développement de ce type de laser continue pour arriver aujourd’hui à des performances élevées. Celles-ci permettent des applications industrielles dans beaucoup de domaines.
Pour le laser XeCl le meilleur donneur de chlore est HCl pour plusieurs raisons :
- sa section efficace d’absorption à 308 nm est faible ; elle est de l’ordre de 10-19 cm2[35]. La concentration en HCl n’affecte pratiquement pas l’énergie de sortie du laser. Ce qui n’est pas le cas pour Cl2 qui a une très forte absorption aux environs de 300 nm[34] - [36] ;
- il a une toxicité moindre que Cl2 ;
- il possède une capacité à se régénérer après dissociation et pulse laser, qui est bien meilleure que les autres donneurs de chlore. Ainsi, il a été observé que 16000 pulses lasers consécutifs ont pu être obtenus sans que l’énergie de sortie en soit affectée[37] ;
- les constantes de vitesse d’excitation vibrationnelle et d’attachement dissociatif par les électrons sont plus favorables pour HCl que pour les autres donneurs de chlore[38]. Ces processus participent efficacement à la formation de XeCl*.
Trois ans plus tard Lorentz et al.[39] réalisent des expériences à haute pression (quelques atmosphères) dans un mélange (Ar/Xe/Cl2) et découvrent une émission centrée à 450 nm qu’ils attribuent à Xe2Cl.
En 1980 un laser Xe2Cl est mis au point[40] - [41]. Ce type de laser apparaît rapidement prometteur car il est susceptible d’être accordable sur une large gamme de longueurs d’onde (30 nm) située dans le visible. Ceci même si des phénomènes d’absorption se produisent du côté des courtes longueurs d’onde et limitent de ce fait l’effet laser à l’aile rouge du spectre de luminescence. Les expériences menées avec Xe2Cl* à l’état solide[42] semblent indiquer que l’état gazeux est plus adapté au développement de ce type de laser bien que les gains mesurés soient corrects à l’état solide. L’état liquide[43] semble beaucoup plus prometteur et se présente comme un laser à colorant idéal bien qu’une mise en œuvre semble complexe et coûteuse. Le laser Xe2Cl n’a pas encore donné lieu à des développements industriels. À la différence de XeCl, le meilleur donneur de chlore est ici CCl4[44] alors qu’aucun effet laser ne se produit si l’on utilise HCl[40].
Quatre molécules peuvent a priori se former dans les mélanges gazeux utilisés pour la formation de chlorures de xénon. Toutes ne se forment pas dans les conditions expérimentales des lasers et leurs rôles peuvent y être différents.
XeHCl a été observé en milieu gazeux. Cependant pour l’instant cette molécule n’a produit que des émissions dans les domaines micro-ondes, radio et lointain infrarouge[45] - [46] bien qu’une émission soit prévue par deux études théoriques à 232 nm[47] et à 129 nm[48]. Notons toutefois que cette molécule proche des agrégats a beaucoup plus de chance d’être stable à l’état solide. Il en va de même pour Xe3Cl dont une émission est prévue théoriquement à 500 nm[49] et qui n’a jamais été observée à l’état gazeux.
XeH a trois émissions recensées à 190 nm[50], 250 nm[51] et 660 nm[52]. Cependant elles ne se sont jamais manifestées dans les spectres laser. Ainsi, on peut penser que les conditions expérimentales n’y sont pas propices à sa formation. À l’inverse, l’ion XeH+ se forme dans les mélanges utilisés dans les lasers. Il y joue un rôle non négligeable dans la cinétique de la formation de XeCl*[53] au travers d’une réaction qui est compétitive avec la création d’ions Xe+ :
HCl+ + Xe → Xe+ + HCl (80 ± 10 %)
HCl+ + Xe → XeH+ + HCl (20 ± 10 %)
La constante de vitesse de l’ensemble des processus est 6,4.10-10 cm3s-1 (± 20 %).
L’ion Xe+ est un précurseur qui joue un rôle capital dans la formation de l’exciplexe.
L’exciplexe XeCl
Structure de la molécule XeCl
Les courbes de potentiel que nous présentons sur la figure 2 résultent de travaux théoriques[48] - [54] - [55], et expérimentaux[56].
On y remarque des états caractéristiques communs à tous les halogénures de gaz rares : un groupe d’états excités B, C et D liés et un groupe d’états plus bas A et X dissociatifs ou faiblement liés. Les états B, D et X ont la symétrie Σ (Λ=1/2) alors que l’état C a la symétrie π (Λ=3/2). L’état A est quant à lui clivé en deux sous-états, l’un de symétrie Σ, A1/2 et l’autre de symétrie π, A3/2.
Avant d’entreprendre une étude détaillée, examinons succinctement les caractéristiques de ces états à partir de considérations très simples.
Le potentiel d’ionisation des gaz rares dans leur état excité le plus bas est voisin de l’affinité électronique des atomes halogènes. Ainsi, les molécules d’halogénures de gaz rares sont formées par une liaison partiellement ionique parce que l’électron excité du gaz rare est partiellement transféré vers l’atome d'halogène. La molécule ainsi formée est donc stable comme c’est le cas des états B, C et D.
Ce transfert d’électron n’a pas lieu lorsqu’on a affaire à des atomes à l’état fondamental : les atomes de gaz rares ne sont normalement pas réactifs et les liaisons interatomiques, quand elles existent, sont essentiellement dues aux forces de van der Waals, toujours de très faible intensité. C’est le cas des états A et X.
États B, C et D
Ces états sont corrélés aux ions Xe+ et Cl− dans leur état fondamental. Le clivage spin-orbite de l’ion Xe+ en deux états 2P3/2 et 2P1/2 est important ; aussi les états B et D qui leur sont corrélés sont-ils très éloignés énergétiquement. Pour les minima des courbes de potentiel qui correspondent pratiquement à la même valeur de la distance internucléaire (re # 0,3 nm), la différence d’énergie mesurée expérimentalement est de l’ordre 9 940 cm−1[57] - [58] - [59]. Celle-ci est donc en accord avec l’énergie de séparation des états Xe+(2P3/2) et Xe+(2P1/2) qui est évalué à 10 574 cm−1.
Les courbes de potentiel des états B et C croisent diabatiquement une courbe de potentiel corrélée à Xe* + Cl à grande distance internucléaire : 7,1 nm expérimentalement[60] et par voie théorique suivant les auteurs 7,19 nm[61] et 6,3 nm[13]. Une investigation théorique plus récente précise ces phénomènes de croisement[62]. Les états B et C qui se confondent à grande distance, croisent successivement deux courbes de potentiel corrélées à Xe* + Cl. La plus basse corrélée à Xe(3P2) + Cl(2P3/2) est croisée 7,25 nm et la suivante corrélée à Xe(3P1) + Cl(2P3/2) est interceptée à 18,68 nm. Comme ce croisement s’effectue à grande distance, le caractère ionique de la liaison de ces états au voisinage de la distance internucléaire d’équilibre re n’est pratiquement pas affecté.
Cette situation est légèrement différente pour l’état D qui croise ces deux courbes de potentiel à une distance beaucoup plus courte[62]. En effet, l’état D croise Xe(3P2) + Cl(2P3/2) à seulement 0,89 nm et Xe(3P1) + Cl(2P3/2) à 1,02 nm.
La distinction entre les états B et C tient au fait qu’ils sont corrélés à des ions Xe+ dont l’orbitale semi-occupée p est dans un plan qui est parallèle à l’axe internucléaire pour l’état B et perpendiculaire à cet axe pour l’état C[63].
Examinons maintenant la question du positionnement énergétique des courbes de potentiel des états B et C. La difficulté provient de leur proximité. Nous avons rassemblé dans le tableau 2 les valeurs de l’intervalle énergétique (EB – EC) entre les deux états. Les données sont très dispersées, en particulier les valeurs calculées sont très éloignées de toutes les valeurs expérimentales. Celles-ci ont été déterminées le plus souvent à partir du rapport des intensités des deux émissions de XeCl* centrées à 308 et 345 mm, corrigé ou non de la participation de la transition (B→A)[64]. La mesure la plus directe est celle de Jouvet et al.[65] : les spectres d’excitation de XeCl* fournissent directement l’écart d’énergie entre les niveaux vibrationnels v’= 0 et v’’= 0 des états respectifs B et C. Cette valeur, 90 cm−1, est proche d’autres mesures réalisées à partir d’études cinétiques[22] - [66] - [67].
EB – EC (cm-1) Méthode Année Référence -1489 Calcul 1977 Goetschalekx et al.[68] -560 Calcul 1978 Hay et Dunning[54] 7 I 1979 Julienne et Krauss[64] 81 I et C 1979 Kolts et al.[55] 128 ± 35 I 1980 Tellinghuisen et Mc Keever[69] -5,4 ± 25 I 1980 Bokor et Rhodes[70] 200 I 1980 Brashears et Setser[71] 230 I 1980 Setser et al.[63] 180 C 1981 Grieneisen et al.[72] 289 I* 1982 Chang[73] 220 ± 40 I 1983 Yu et al.[74] 85 C 1984 Inoue et al.[66] 0 C 1984 Lorents[75] -22 C 1985 Le Calvé et al.[76] > 50 I** 1986 Fajardo et Apkarian[77] 230 ± 40 I 1987 Lo et Zheng[56] 90 ± 2 Absorption 1989 Jouvet et al.[65] 98 +30-40 I et C 1990 Quiñones et al.[67] 118 ± 40 I 1992 Asselman et al.[21]
Tableau 2 : Ecart d’énergie (EB – EC) entre les états B et C de XeCl.
I : mesure déduite de la valeur du rapport des intensités des émissions de XeCl centrées à 308 et 345 nm (cf. § 3-1-1).
C : mesure déduite d’une étude cinétique fournissant les constantes de couplage entre ces deux états.
* : l’émission à 345 nm n’est pas corrigée de la contribution XeCl (B → A).
** : XeCl est à l’état solide.
Le positionnement de l’état B par rapport à l’état C est justifié théoriquement par la prise en compte de l’interaction de configuration entre les états de caractères ioniques et covalents de même symétrie[55] - [70] - [78] - [79]. Dans un état 2Σ (comme les états B et X), une orbitale simplement occupée est située plus près d’une orbitale d’un autre atome de telle manière que l’interaction ou l’échange de charges entre deux atomes sont plus importants et plus faciles que dans un état 2π (comme les états C et A3/2) où l’orbitale simplement occupée est perpendiculaire à l’axe moléculaire et loin d’un autre atome. La correction introduite par ce phénomène dans les valeurs de l’énergie est donc beaucoup plus importante pour les états Σ que pour les états π[78]. La prise en compte de cette interaction de configuration, augmente de manière beaucoup plus significative l’énergie de l’état B que celle de l’état C. D’où le positionnement observé sur les courbes de potentiel de la figure 2.
États X et A
Les états les plus bas sont corrélés aux atomes de xénon et de chlore à l’état fondamental.
En raison du clivage spin-orbite de l’atome de chlore (881 cm−1)[80] en deux états (2P3/2) et (2P1/2), l’état A se scinde en deux sous-états. Cependant, l’effet du couplage spin-orbite est ici nettement plus faible que dans le cas de l’ion Xe+. À grande distance internucléaire, un écart énergétique de 882 cm−1 entre A1/2 et A3/2 a été mesuré expérimentalement à l’état solide dans une matrice de néon[81]. Cette valeur est donc très proche de la séparation énergétique des états Cl(2P3/2) et Cl(2P1/2). Ce qui confirme les hypothèses théoriques de corrélations d’états. On notera qu’à grande distance l’état A3/2 se confond avec l’état X. Ce résultat est confirmé expérimentalement par Becker et al.[82] qui ont construit des potentiels d’interaction de 35Cl(2P3/2 et 2P1/2) et de Xe(1S0) à partir de l’analyse de diffusions quasi-élastiques dans des collisions produites à partir de faisceaux croisés.
Contrairement à certains autres halogénures de gaz rares, XeCl possède un état fondamental non dissociatif. Ce caractère liant a été mis en évidence expérimentalement bien avant les travaux théoriques dans des études sur des molécules XeCl à l’état solide dans des matrices d’argon à 20K[59] et plus tard à l’état gazeux[58] - [60]. Les forces de van der Waals qui interviennent dans la liaison entre atomes[83] ne sont pas suffisamment intenses dans le cas de l’état X pour expliquer la présence d’un puits de potentiel qui même faible (la profondeur est de l’ordre de kT), peut contenir entre 12 et 20 niveaux vibrationnels (voir tableau 3). L’augmentation relative de l’énergie de liaison dans l’état X par rapport à celle de l’état A peut s’expliquer là encore par la prise en compte de l’interaction de configuration[84]. Nous allons voir que l’état A est lui aussi très légèrement lié. Son énergie de liaison est deux fois moins élevée que celle de l’état X.
Tableau 3 : Déterminations expérimentales du nombre de niveaux vibrationnels contenu dans le puits de l’état X.
Constantes spectroscopiques de XeCl
L’énergie Ev’j’M d’un état M sur un niveau vibrationnel v’ possédant le nombre quantique de rotation j’ est :
Ev’j’M = Te(M) + EVib(M) + ERot(M)
où Te(M), EVib(M) et ERot(M) désignent respectivement les énergies électronique, vibrationnelle et rotationnelle de la molécule.
Structure électronique
Les principales caractéristiques des états électroniques M sont habituellement l’énergie de dissociation De, la distance inter-atomique re et l’énergie du fonds du puits EM. Pour XeCl, les différentes valeurs publiées de ces grandeurs sont rassemblées dans les tableaux 4, 5 et 6. Elles ont été déterminées par voie théorique ou expérimentale pour l’isotope 35Cl à l’état solide ou gazeux.
États Réf
X A B C D Becker et al.[82] 280 ± 7 % 129 ± 7 % Hay et Dunning[54] 33 957 33 392 33 634 Last et George[49] 36 699 Sur et al.[86] 281 ± 10 36 553 37 148 Tellinghuisen et al.[60] 255 ± 10 36 540 Tellinghuisen[88] 281,1 ± 0,7 Haberland[89] 154 Aquilanti et al.[84] 161 Flannery[13] 225 Johnson et al.[90] 35 459
Tableau 4 : Énergies de dissociation De en cm-1.
Il est nécessaire d’analyser en détail l’ensemble de ces résultats compte tenu des méthodes différentes utilisées et du fait que nous y avons aussi inclus des mesures anciennes.
Énergies de dissociation
Le tableau 4 fait apparaître une grande disparité entre le nombre de déterminations pour chaque état. Une analyse statistique des résultats est impossible pour les états A, C et D qui n’ont fait l’objet au maximum que de trois mesures. Pour l’état B, les quatre valeurs ne sont pas cohérentes entre elles et il est difficile en première approche de mettre en doute l’une d’elles. Notons simplement l’accord sur l’ordre de grandeur.
Pour l’état X, nous disposons de six mesures dont deux sont statistiquement éloignées des autres. Celle de Flannery[13] est une estimation théorique ancienne et peu précise. Celle de Tellinghuisen et al.[60] est une première détermination expérimentale faite en 1976. Sept ans plus tard[88] la même équipe corrigera cette valeur et la situera très proche des estimations les plus récentes. Les quatre valeurs restantes nous paraissent les seules fiables. On peut alors déterminer statistiquement un intervalle de confiance, pour lequel on se fixe arbitrairement comme seuil la valeur classique de 5 %. On trouve alors que De doit se trouver compris (avec une probabilité de 95 %) entre 278,3 cm−1 et 285,3 cm−1. Malgré le caractère étroit de cet intervalle qui correspond à une fluctuation de 1,3 % autour de 281,5 cm−1, il faut insister sur l’importance d’une détermination plus précise. En effet, parmi les déterminations retenues figurent deux mesures avec une forte incertitude[82] - [86], et une troisième où l’auteur ne la signale pas[84]. De la valeur de De de l’état X, dépend le nombre de niveaux vibrationnels contenus dans le puits et qui fixe le nombre de transitions lié → lié qu’il est possible d’obtenir. Ce résultat est fondamental pour mieux connaître la spectroscopie des lasers XeCl.
En conclusion nous dirons qu’il est nécessaire de préciser les valeurs de De pour tous les états de XeCl. D’autant plus que parmi les quelques résultats relatifs aux états A, B, C et D figurent des travaux anciens qui pour d’autres grandeurs fournissent des mesures discutables comme nous allons le voir.
Distances atomiques d’équilibre
États Réf
X A B C D Adrian et Jette[91] 3,44 Becker et al.[82] 3,23 4,1 Hay et Dunning[54] 3,22 3,14 3,18 Krauss[78] 3,227 Fajardo et Apkarian[77] 3,14 Sur et al.[86] 3,23 3,007 2,922 Tellinghuisen et al.[60] 3,18 2,9374 Yu et al.[74] 3,074 Haberland[89] 4,05 Aquilanti et al.[84] 3,23 4,09 Ewing et Brau[61] 2,9 Adams et Chabalowski[48] 3,17 3,08 3,12
Tableau 5 : Distance interatomique d’équilibre re en Ǻ.
L’examen du tableau 5 nous indique qu’il y a peu de mesures pour les états A, C et D. Mais cette fois-ci, les valeurs sont proches les unes des autres. Bornons-nous à en donner les valeurs moyennes : pour l’état A nous avons 0,408 nm, pour l’état D 0,307 nm et pour l’état C 0,311 nm.
Pour l’état X, la détermination théorique ancienne d’Adrian et Jette[91] est statistiquement éloignée des autres. Sans en tenir compte nous trouvons pour re l’intervalle de confiance au seuil de 5 % suivant : 0,318 nm < re < 0,326 nm.
Remarquons que la valeur de Tellinghuisen et al.[60] est à la limite de l’intervalle. Nous avons déjà mis en doute un résultat de cette étude au paragraphe précédent. Si nous n’en tenons pas compte, on s’aperçoit qu’alors les trois autres auteurs annoncent une valeur identique de 0,323 nm. Enfin, nous constatons que cette publication fournit à nouveau une valeur statistiquement éloignée des autres pour la valeur de re qui concerne l’état B. Il en est de même pour celle de Ewing et Brau[61] qui est l’étude la plus ancienne des halogénures de gaz rares. Ses conclusions reposent sur l’analogie des gaz rares excités avec les métaux alcalins. Les résultats ne fournissent donc que des estimations d’ordres de grandeur. Ces deux valeurs seront écartées pour donner un intervalle de confiance au seuil de 5 % pour la distance interatomique relative à l’état B : 0,299 3 nm < re < 0,331 9 nm.
La situation des déterminations de re est satisfaisante même si des mesures pour l’état C et l’état D pourraient apporter une information intéressante.
Énergies du fonds du puits
Tableau 6 : Énergie du fond du puits Ei en cm-1.
Le tableau 6 nous montre qu’il y a très peu d’informations pour les états X, A et D. Pour l’état X, il faut préciser que Sur et al.[86] ont pris arbitrairement le fonds du puits de X comme origine de leur échelle d’énergie. Il ne s’agit donc pas d’une mesure directe. De ce fait, l’état X comme l’état A n’ont fait l’objet que d’une seule étude, celle de Aquilanti et al.[84]. Pour l’état D, il n’y a que deux déterminations assez différentes. Pour l’instant, on ne peut pas apporter de conclusion pour D mais nous y reviendrons à la fin de ce paragraphe.
Le positionnement de l’état B et de l’état C pose problème comme nous l’avons vu au chapitre précédent. L’analyse suivante va nous permettre de préciser la situation.
Intéressons-nous tout d’abord à l’état B qui a le plus retenu l’attention des chercheurs jusqu’à présent. Deux des mesures sont statistiquement éloignées des autres. Outre la publication d’Ewing et Brau[61] dont nous avons déjà parlé, figure parmi les déterminations douteuses, les travaux théoriques anciens de Hay et Dunning[54] sur lesquels nous reviendrons. Sans tenir compte de ces valeurs, les travaux expérimentaux fournissent un intervalle de confiance au seuil de 5 % qui est très étroit : de 32 380,1 cm−1 à 32 415,3 cm−1.
Par contre pour l’état C, en première approche, aucune conclusion ne peut être tirée statistiquement compte tenu du faible nombre de mesures. Cependant, une autre analyse va nous permettre d’y voir plus clair malgré le caractère non concordant des valeurs du tableau 6. En effet, comme nous l’avons au paragraphe 1.1 le positionnement de l’état C par rapport à l’état B a donné lieu à beaucoup de publications.
Une analyse statistique des valeurs du tableau 2 permet de proche en proche d’arriver à un intervalle de confiance au seuil de 5 % qui est le suivant : 76,8 cm−1 < (EB – EC) < 100,2 cm−1. Seules quatre mesures appartiennent à cet intervalle. Il s’agit de la détermination directe de Jouvet et al.[65] et des trois valeurs déduites des études cinétiques[55] - [66] - [67]. D’autre part, notons que l’estimation ponctuelle donne 88,5 cm−1 et que la seule mesure à être en accord avec elle (compte tenu de l’erreur absolue annoncée) est celle de Jouvet et al.[65] qui vaut (90 ± 2 cm−1). Notre étude statistique confirme donc les conclusions du paragraphe 1.1.
Revenons à l’énergie du fond du puits de l’état C. Si on considère les intervalles de confiance énoncés plus haut pour l’état B et la différence d’énergie (EB – EC), on peut en déduire un intervalle pour EC : 32 279,9 cm−1 < EC < 32 338,4 cm−1.
Dans ces conditions, seule la valeur de Jouvet et al.[65] du tableau 6 est compatible avec cet intervalle. Parmi les trois déterminations douteuses on retrouve celle de Hay et Dunning[54] qui donnaient déjà une valeur imparfaite pour EB. Une autre étude théorique ancienne effectuée par Clugston et Gordon[92] sort elle aussi de cet intervalle. Il en est de même pour les travaux expérimentaux menés à l’état solide par Fajardo et Apkarian[77].
Revenons maintenant à l’état D et calculons la moyenne des deux valeurs du tableau 6. On trouve 43 838,45 cm−1. L’écart énergétique avec l’état B est alors de l’ordre de 11 400 cm−1. Par ailleurs, Shostak et Strong[57] ont déterminé expérimentalement la différence d’énergie entre les états D et B. Ils ont trouvé 9 900 cm−1. La différence entre ces valeurs de (EB – ED) est très nette. Notons que si on se limite aux travaux de Sur et al.[86], l’écart d’énergie entre les états D et B devient de l’ordre de 9 950 cm−1 qui est proche de la détermination de Shostak et Strong[57]. Cette remarque met en doute à nouveau les travaux théoriques de Hay et Dunning[54] pour lesquels (EB – ED) serait 10 888 cm−1.
En conclusion par rapport à la structure électronique, il apparaît qu’un certain nombre d’études anciennes posent problème quant à quelques-uns de leurs résultats[13] - [54] - [60] - [61] - [92]. D’un autre côté, les travaux menés à l’état solide par Fajardo et Apkarian[77] ne sont pas toujours en accord avec ceux effectués à l’état gazeux. Par ailleurs, les études théoriques récentes n’ont pas résolu tous les problèmes et des différences notables avec les résultats des expériences demeurent[48] - [49].
À ce niveau d’analyse, on peut revenir en arrière et reconsidérer le rôle de publications douteuses[54] - [60]. Le retrait des valeurs de Hay et Dunning[54], réduit à une détermination les valeurs de De pour l’état C et l’état D, et rend homogènes les trois autres valeurs relatives à l’état B. Parmi celles-ci on retrouve quand même celle de Tellinghuisen et al.[60] qui pose problème pour d’autres grandeurs. L’énergie De pour l’état B a alors une valeur moyenne de 36 184 cm−1.
Structure vibrationnelle
L’énergie de vibration du niveau v’ de l’état M vaut :
EVib(M) = ωe (v’+1/2) - ωexe (v’+1/2)2
où ωe et (ωexe) désignent respectivement la fréquence fondamentale vibrationnelle et la constante d’anharmonicité. Leurs différentes déterminations sont rassemblées dans les tableaux 7 et 8.
Fréquences fondamentales vibrationnelles
Les valeurs de ωe sont rassemblées dans le tableau 7.
États Réf
X B C D Brau et Ewing[94] 210 Hay et Dunning[54] 188 188 189 Jouvet et al.[65] 27 ± 1 193 ± 1 204 ± 1 Kvaran et al.[95] 194,235 Sur et al.[86] 26,22 194,75 204,34 Tellinghuisen et al.[60] 26,27 (± 0,55) 195,17 (± 0,31) Tamagake et al.[79] 195,6 Ault et Andrews[59] 50 ± 10 Fajardo et Apkarian[77] 188 Huber et Herzberg[93] 195,2 Clugston et Gordon[92] 187 Le Calvé et Gürtler[96] 210 Adams et Chabalowski[48] 195 198 Golde[97] 205 ± 12
Tableau 7 : Valeurs de ωe en cm-1.
Pour les états X, C et D, il n’y a que quatre déterminations et aucune mesure ne peut être considérée comme statistiquement éloignée des autres, même si l’on constate des disparités. Nous reviendrons plus loin sur les états C et D.
Pour l’état B, nous disposons de neuf déterminations. Une analyse statistique conduit à un intervalle de confiance au seuil de 5 % qui est très étroit : 194,7 cm−1 < ωe < 195,4 cm−1.
On constate que six valeurs du tableau 7 n’en font pas partie. Trois d’entre elles le sont très nettement. Il s’agit de publications anciennes dont deux (Hay et Dunning[54] et Brau et Ewing[94]) ont déjà fait l’objet de notre critique au paragraphe précédent. Pour ce qui concerne la troisième Golde[97], remarquons qu’elle est fondée sur la même méthode que celle utilisée par Brau et Ewing[94]. Les trois autres mesures qui se situent hors de l’intervalle sont plus récentes. Celle de Kvaran et al.[95] a été réalisée à l’état solide. Comme Fajardo et Apkarian[77] dans le paragraphe précédent, ils observent des différences notables avec l’état gazeux. Par contre, plus surprenants sont les désaccords de Jouvet et al.[65] et Tamagake et al.[79] qui ont fourni par ailleurs de bons résultats. Remarquons enfin que parmi les valeurs qui appartiennent à l’intervalle, figurent autant de travaux théoriques (Adams et Chabalowski[48] et Huber et Herzberg[93]) qu’expérimentaux (Tellinghuisen et al.[60] et Sur et al.[86]).
En conclusion, on s’aperçoit qu’ici les travaux de Tellinghuisen et al.[60] donnent des résultats très satisfaisants à la fois sur l’état B et sur l’état X.
Pour ce qui est de l’état C, on ne peut que constater qu’il n’y a que des références qui ont donné des résultats contestables auparavant (Hay et Dunning[54], Fajardo et Apkarian[77] et Clugston et Gordon[92]) dont celle de Jouvet et al.[65] qui, on vient de le voir, est éloignée des autres mesures dans le cas de l’état B.
Pour l’état D, faisons la remarque que le retrait de la détermination de Hay et Dunning[54] rend plus homogène les trois autres valeurs.
Finalement il apparaît nécessaire de préciser les valeurs de ωe pour les états X, C et D. L’intérêt principal de cette clarification serait une meilleure résolution de la structure vibrationnelle de la transition utilisée dans le laser, laquelle passe par une meilleure connaissance de l’état X. D’autre part la structure de l’état C est importante car elle joue un rôle fondamental au niveau de la cinétique du laser.
Constantes d’anharmonicité
À l’examen du tableau 8, aucune conclusion ne peut être avancée en ce qui concerne les valeurs des constantes d’anharmonicité pour les états X, C et D.
Tableau 8 : Valeurs de ωe.xe en cm-1.
Par contre pour l’état B nous disposons de six mesures. L’intervalle de confiance au seuil de 5 % est le suivant :
0,532 cm−1 < ωe.xe < 0,669 cm−1.
Encore une fois celle de Jouvet et al.[65] est statistiquement éloignée des autres et les auteurs ne peuvent expliquer cette différence. Notons que les travaux de Hay et Dunning[54] fournissent cette fois-ci des prévisions correctes. Il en est de même pour l’étude de la structure vibrationnelle réalisée par Tellinghuisen et al.[60].
En conclusion il apparaît nettement que la structure vibrationnelle des états électroniques connus de XeCl doit être précisée même si l’état B a été plus étudié et mieux caractérisé.
Structure rotationnelle
L’énergie de rotation est donnée par l’expression :
Erot(M) = B’.K’ef – D’.(K’ef)2,
où K’ef = j’(j’+1) ± (1/2).δ(j’+1/2) ;
B’ et D’ sont respectivement la constante rotationnelle et la première constante de distorsion centrifuge. Leurs valeurs sont données dans les tableaux 9 et 10. δ est un paramètre qui vaut 2,0 pour l’état B Quiñones et al.[67] et 0,4 pour l’état X[98].
Tableau 9 : Valeurs de B’ en cm-1.
La structure rotationnelle est donc mal connue. On notera cependant la concordance des quelques mesures relatives à B’.
Tableau 10 : Valeurs de D’ en cm-1.
Voies de formation
Lorsqu’ils se trouvent dans des états métastables de configuration np5(n+1)s1, (n=5 pour le xénon), les gaz rares possèdent des propriétés de polarisabilité et de diffusion élastique analogues à celles des métaux alcalins[100]. L’électron de valence s du gaz rare excité a une énergie de liaison voisine de celle du métal alcalin qui le suit dans la classification périodique des éléments. Dans un certain nombre de publications anciennes[61] - [97] - [101] - [102], cette analogie valable uniquement pour les gaz rares les plus lourds, est mise à profit pour étudier le comportement de ces gaz avec les donneurs d’halogènes. On sait en particulier que les alcalins possèdent une bonne affinité chimique vis-à-vis des halogènes ; il doit en être de même des atomes excités de gaz rares. Expérimentalement la section efficace de collision des états métastables des gaz rares avec les halogènes est remarquablement proche de celle des alcalins avec les halogènes[101] - [102] - [103]. Ainsi, le xénon excité possède une structure électronique voisine de celle du césium et on peut comprendre qu’il puisse réagir avec un donneur de chlore pour former XeCl*.
Des analyses plus récentes montrent toutefois des différences notables entre métaux alcalins et gaz rares excités. Elles se manifestent au niveau de la symétrie des molécules produites. D’autre part, le nombre d’états d’halogénures de gaz rares est plus grand que celui des sels produits à partir des métaux alcalins. Cela est dû au clivage spin-orbite des atomes et des ions de gaz rares.
La première condition pour produire du XeCl est donc de rendre le xénon réactif. Pour cela, il faut soit l’exciter, soit l’ioniser, soit les deux à la fois. Plusieurs méthodes d’excitation extérieures ont été employées. Les plus courantes sont les décharges électriques[33], les faisceaux d’électrons[44], l’excitation laser[104], les micro-ondes[105] et les particules α[21].
À partir du moment où l’excitation n’est pas sélective, la formation de XeCl* peut se faire suivant de nombreuses voies. Leur importance relative varie avec les conditions expérimentales, principalement la pression, le moyen d’excitation et surtout le donneur d’halogène. Lorsque les mélanges sont ternaires les processus de création de XeCl se compliquent. Malgré tout, l’addition d’un gaz tampon présente de nombreux avantages. En effet, les autres gaz rares sont moins coûteux que le xénon mais surtout ils possèdent (ainsi que leurs espèces excitées et leurs ions) des sections efficaces d’absorption à 308 nm nettement inférieures à celles des mêmes espèces du xénon. Ainsi, le gaz tampon peut-il être utilisé en de très fortes proportions sans trop altérer l’énergie de sortie du laser. Dans ces conditions, les proportions de xénon et de HCl sont limitées à la juste dose nécessaire pour produire une quantité suffisante d’exciplexe. Le rôle essentiel du gaz tampon est de transférer aux atomes de xénon l’énergie d’excitation qui lui est communiquée. Ce transfert peut être considéré comme instantané et il est très efficace. Il peut se traduire par l’excitation ou l’ionisation du xénon ou par la formation d’un ion RgXe+[6]. Chacune de ces espèces peut ensuite réagir avec le donneur de chlore pour former XeCl*. D’un autre côté, la formation d’espèces neutres RgXe ne semble pas être importante[7].
Il existe donc deux voies principales de formation d’exciplexes :
- des collisions entre d’une part des molécules de donneur de chlore et d’autre part des atomes ou des molécules de xénon, l’une des espèces au moins étant excitée. Le gaz tampon peut parfois être un partenaire de ce type de réaction ;
- des recombinaisons d’ions. Le gaz tampon est presque toujours impliqué dans ces réactions.
Notons que la formation de XeCl* est extrêmement efficace puisque Konovalov et al.[106] ont observé une émission de XeCl alors que le xénon ne se trouvait qu’à l’état de traces (0,2 %) dans du krypton.
Avant d’étudier en détail les deux mécanismes principaux de formation de l’exciplexe, nous allons parler d’une troisième possibilité qui présente un intérêt plus limité pour les lasers. Toutefois, elle permet de mieux comprendre le mécanisme réactif. Il s’agit de la voie photoassociative.
Voie photoassociative
La synthèse de XeCl* se produit lorsque l’on excite un mélange de xénon et de chlore (Cl2) au moyen d’un laser émettant entre 304 et 312 nm[104]. Deux réactions sont alors induites[107] :
- une excitation d’un état électronique isolé d’un atome de xénon ou d’une molécule suivie d’une collision réactive ;
- une vraie réaction induite par laser : interaction simultanée d’une paire en collision et un ou deux photons qui produisent un état intermédiaire qui fournit ensuite les produits sans qu’intervienne de collision.
Dans ce dernier cas, il se forme un complexe transitoire[108] (Xe-Cl2)* dans l’état (1Πu)[109]. Dès lors deux voies de dissociation sont possibles à partir du moment où un photon est absorbé par la paire Cl-Cl ou par la paire Xe-Cl de (Xe-Cl2)* dans l’état (1Πu)[109] - [110] :
Xe-Cl2(1Πu) + hν → Xe-Cl2(1Πg) → Xe+Cl2− → XeCl(B,C) + Cl
Xe-Cl2(1Πu) + hν → Xe-Cl(X)-Cl + hν → Xe-Cl(B)-Cl → XeCl(B) + Cl
La constante de vitesse de la réaction a été mesurée en considérant le photon comme troisième partenaire. Elle vaut 6.10-29 cm6.s-1[111].
Des résultats analogues ont été obtenus avec d’autres donneurs de chlore dont HCl et CCl4.
Dans tous les cas, les molécules XeCl(B,C) sont toujours produites dans des états qui comportent une forte excitation vibrationnelle.
Voie collisionnelle
Il existe de nombreux processus dont l’importance dépend de la nature et de l’excitation des espèces en collision. La principale demeure dans tous les cas le harponnage qui résulte toujours des collisions binaires.
Collisions de type « harpon »
Ces réactions font intervenir le donneur de chlore à l’état fondamental et un atome excité du xénon, sur les premiers niveaux 6s, Xe* et sur des niveaux plus élevés Xe** dont les niveaux 6p.
Mécanisme du « harponnage »
D’une manière générale, ces réactions permettent de décrire le résultat des collisions entre des atomes de gaz rares (Rg) et des donneurs d’halogènes (RX), où X est l’atome halogène et R un radical moléculaire[112]. Les produits des réactions dépendent fortement du gaz rare et du donneur d’halogène impliqués. Dans notre cas où Rg=Xe et X=Cl, la nature des produits suit cette règle[55] - [113]. Cette collision peut même ne pas fournir du tout d’halogénure de gaz rare dans certains cas. Pour bien comprendre ce qu’il se passe, il faut examiner en détail le processus de collision entre Rg et RX[55].
L’atome Rg et la molécule RX suivent lors de leur approche le potentiel adiabatique le plus bas et la réaction se déroule par le mécanisme d’orbite contrôlée de croisement de courbe ionique-covalent. Les réactifs (Rg et RX) approchent sur une surface diabatique covalente. Il se forme alors un complexe Rg*…RX à une distance internucléaire assez grande. Son potentiel est V(Rg, RX). Lorsque la distance devient suffisamment faible, il se peut que V(Rg, RX) croise une surface de potentiel ionique V(Rg+…RX−). Le croisement peut se manifester par le transfert d’un électron de Rg vers RX. On dit que l’on a affaire à un mécanisme de harponnage. Dans ce cas, les atomes continuent sur la nouvelle surface. Ce qui conduit à la diffusion réactive et à la formation de RgX*.
La figure 3 nous montre le processus de création de XeCl* qui fait intervenir Rg=Xe et X=Cl. Après son transfert, l’électron occupe une orbitale antiliante de RCl. En présence de Xe+, RCl− se divise en R et Cl−. Les ions Xe+ et Cl− se recombinent ensuite pour former XeCl dans les états B, C et D car il n’y a pas de nouvelle force entre Cl− et R. L’excitation vibrationnelle du produit XeCl* est toujours importante. Au total, tout se passe comme si on avait la réaction :
Xe* + RCl → XeCl*(B,C,D) + R
avec la constante de vitesse kMX.
Cependant, des réactions compétitives à la formation de XeCl* se manifestent avant ou après le croisement. Elles correspondent aux interactions des potentiels V(Rg+, RX−) et V(Rg + RX*).
D’une manière générale, cette situation se produit lorsque la surface ionique est coupée par des surfaces covalentes où RX est dans son état excité le plus bas. La distribution de sortie dépend du nombre et de la nature des voies de sortie qui sont possibles à la suite de la collision[112] - [114]. La plus fréquente se manifeste à l’intersection des surfaces de potentiel par un transfert d’énergie électronique qui peut amener une dissociation de l’accepteur excité :
Rg* + RX → (Rg+…RX−) → Rg(B,C,D) + RX*
avec la constante de vitesse kET.
Rg* + RX → (Rg+…RX−) → Rg + R + X
avec la constante de vitesse kD.
Cette voie tend à devenir moins importante à mesure que la complexité de RX croît.
Il est aussi possible que le transfert se fasse sur un état qui n’est pas corrélé à l’ion RX− mais à des niveaux de Rydberg très hauts dans la molécule neutre et se trouvant juste en dessous de la limite d’ionisation. Les facteurs critiques régulant les rapports de branchement sont les énergies des potentiels corrélés avec l’ion moléculaire (VI), le groupe de Rydberg proche de l’ionisation (VII) ou un atome excité initial (VIII). L’importance de ces voies croît avec la profondeur du puits de V(Rg+, RX−).
Quand les niveaux d’énergie asymptotiques à grande séparation se trouvent dans l’ordre VI > VII > VIII et que les potentiels (VII) sont attractifs, le premier croisement évité rencontré lorsque les atomes réagissant s’approchent favorise en sortie (VII) plutôt que le potentiel anionique (VI). Vu que (VII) a un cœur cationique qui reste fortement lié, il conduit préférentiellement à un transfert d’excitation. C’est la réaction d’excitation dissociative :
Rg* + RX → Rg + R* + X ou Rg + R + X*
avec la constante de vitesse kDE
Si VIII > VII à longue distance, des voies d’ionisation de Penning ou d’ionisation associative peuvent être ouvertes[112] :
- ionisation de Penning : Rg* + RX → Rg + RX+ + e−
avec la constante de vitesse kPI
- ionisation associative : Rg* + RX → (RgRX)+ + e−
avec la constante de vitesse kAI.
Dans (VI) la liaison avec un atome halogène est en principe faible et le transfert atomique est favorisé entre Rg et R. Ce potentiel conduit donc à la formation de l’exciplexe.
Il existe donc a priori cinq voies compétitives à la création de RgX. Pour XeCl* un atome de xénon excité entre en collision avec un donneur de chlore. Ces cinq réactions ont toutes été observées[114] pour divers donneurs de chlore. Pour chiffrer la proportion de la production d’exciplexe, il est d’usage de définir le rapport de branchement. Il indique le taux de formation de XeCl ; on le note ΓXeCl :
ΓXeCl = kMX / (kMX + kAI + kPI + kET + kDE + kD)
Des mesures de ΓXeCl ont été réalisées pour plusieurs donneurs de chlore et essentiellement pour les états 6s et 6p du xénon.
Xe(6s ou 6p) + RCl → produits
avec la constante de vitesse kQ
kQ est la constante de vitesse totale et elle vaut :
kQ = kMX + kAI + kPI + kET + kDE + kD .
État du xénon kQ × 10-10 Références 3P2 ou (6s[3/2]2) (10 ± 2) Maeda et al.[115] 3P2 ou (6s[3/2]2) 7,2 Velazco et al.[114] 3P2 ou (6s[3/2]2) (7,0 ± 0,5) Berman[116] 3P1 (7,9 ± 0,9) Lorents[75] 1P1 (7,6 ± 0,7) Lorents[75] (6p[1/2]0) (14,6 ± 0,2) Setser et Ku[107] (6p[1/2]0) (17,9 ± 0,2) Bruce et al.[117] (6p[1/2]2) (14,5 ± 0,2) Setser et Ku[107] (6p[1/2]2) (15,5 ± 0,2) Bruce et al.[117] (6p[5/2]2) (13,3 ± 1,0) Ku et Setser[118] (6p[5/2]2) (12,8 ± 0,3) Bruce et al.[117] (6p'[3/2]2) (18,6 ± 0,5) Bruce et al.[117] (6p'[1/2]0) (21,9 ± 1,0) Bruce et al.[117] (7p[5/2]2) (30,7 ± 1,9) Bruce et al.[117] (7p[1/2]0) (29,5 ± 0,8) Bruce et al.[117] (7d[1/2]1) (9,2 ± 0,5) Bruce et al.[117]
Tableau 11 : Constantes de vitesse totales en cm3s-1 pour les collisions harpon entre Xe* et Cl2. Ici ΓXeCl = 1.
Les résultats concernant le chlore Cl2, CCl4 et HCl(v=0) sont rassemblées dans les tableaux 11,12 13. ΓXeCl est posé égal à 1 par Setser et Ku[107] dans le cas où le donneur de chlore est Cl2. Cette décision est justifiée par le fait que pour Xe* + Cl2 nous avons VII > VI > VIII. Ce qui impose selon Simons[112] que la voie correspondant au transfert d’excitation est très improbable.
État du xénon kQ × 10-10 ΓXeCl Références 3P1 ou (6s[3/2]1) 6,2 0,01 Setser et al.[63] 3P2 ou (6s[3/2]2) (7 ± 2) Maeda et al.[115] 3P2 ou (6s[3/2]2) 5,6 0,01 Setser et al.[63] et Velazco et al.[114] 3P2 ou (6s[3/2]2) 5,6 <0,02 Kolts et al.[55] 1P1 4,62 Chen et Setser[119] 1P1 7 ≈0 Lorents[75] (6p[1/2]0) (8,3 ± 0,5) 0,80 ± 0,15 Ku et Setser[118] (6p[3/2]2) (8,0 ± 0,5) 0,60 ± 0,15 Ku et Setser[118] (6p[3/2]2) (6,5 ± 0,2) Setser et Ku[107] (6p[5/2]2) (8,0 ± 0,5) 0,40 ± 0,15 Ku et Setser[118] 5d[3/2] (15,6 ± 1,5) 0,48 Lorents[75] Ensemble des états 6p 5 Wren et al.[120] Ensemble des états 6p 5,6 0,60 Levin et al.[121]
Tableau 12 : Constantes de vitesse totales en cm3s-1 et ΓXeCl pour les collisions harpon entre Xe* et HCl (v = 0).
Une première analyse des tableaux 11 à 13 montre que les résultats sont en assez bon accord entre eux lorsque plusieurs mesures ont été effectuées pour une même réaction. Nous constatons que la plupart des collisions ont eu leur constante de vitesse qui n’a été mesurée qu’une seule fois. D’autre part, à de rares exceptions près, ces déterminations de kQ et ΓXeCl se limitent aux états excités les plus bas du xénon atomique. Ceci indique l’intérêt de nouvelles mesures afin de confirmer les résultats expérimentaux disponibles et d’estimer le rôle des autres états qui ne manquent pas de se former si l’on utilise, comme c’est le cas dans les lasers, des moyens d’excitation non sélective.
Tableau 13 : Constantes de vitesse totales en cm3s-1 et ΓXeCl pour les collisions harpon entre Xe* et CCl4.
Un résultat important pour les lasers XeCl apparaît en première analyse. Xe(6s) + HCl(v=0) ne produit pas de XeCl. Or d’après les estimations de Kannari et al.[123] une proportion de 5 % de la formation d’exciplexes se fait dans les lasers par la voie du harponnage. De plus, les états Xe(6p) en produisent 2,5 %. Nous nous proposons maintenant d’expliquer ce résultat.
États de départ : Xe(6s)
Le chlore moléculaire réagit de manière efficace avec ces états du xénon. Il ne faut donc pas négliger cette réaction dans la cinétique du laser XeCl, compte tenu du fait que Cl2 se forme dans le mélange gazeux (figure 1).
La réaction avec CCl4 est beaucoup moins rapide qu’avec Cl2, d’un ordre de grandeur, mais elle est quand même efficace. Cette réaction sera donc importante dans la cinétique des lasers Xe2Cl.
Si le donneur de chlore est HCl, la situation est plus complexe. Deux situations se présentent :
. HCl à l’état fondamental est sur le niveau vibrationnel v=0. Les valeurs de kD sont très proches quel que soit l’état de départ du xénon ; le rapport de branchement pour les états 6s est très faible. La contribution apportée par ces états à la formation de XeCl* est négligeable. De plus, les réactions compétitives qui sont présentes se produisent avant le croisement des courbes de potentiel V(Xe*+HCl) et V(Xe++HCl−)[120]. Le quenching de Xe(6s) par HCl est donc important dans la cinétique du laser. Il détruit des états du xénon susceptibles de former des exciplexes.
. HCl à l’état fondamental est sur le niveau vibrationnel v=1. Pour l’état Xe(3P2), Chang[73] montre qu’il y a une augmentation très nette du taux de production de XeCl. La constante de vitesse de formation de XeCl qu’il mesure a une valeur minimale de 2.10-10 cm3s-1 et ΓXeCl = 35 %. Notons que la première estimation faite par Levin et al.[121] et fondée sur une analogie, annonçant 6.10-11 cm3s-1 et ΓXeCl = 11 % pour cette réaction, ne soit plus être prise en compte après les mesures directes de Chang. Si l’excitation vibrationnelle de HCl croît, le taux de formation de XeCl doit encore certainement être efficace. Il n’y a pas de mesure directe disponible mais des estimations sont proposées par des équipes de modélisation s’appuyant sur des analogies. Pour v=2 deux valeurs des constantes de formation ont été trouvées dans la littérature : 5,6.10-10 cm3s-1[124] et 2,0.10-10 cm3s-1[125].
Pour d’autres auteurs, c’est l’ensemble des niveaux vibrationnels qui sont pris en compte. Ainsi pour v≥1, Kannari et al.[126] proposent comme constante de formation 5,6.10-10 cm3s-1 et ΓXeCl = 26 %. Des expériences sont donc nécessaires pour préciser cette partie cinétique qui joue un rôle fondamental dans les lasers comme l’ont prévu Kannari et al.[123].
États de départ : Xe(6p)
Les réactions de formation de XeCl sont d’une manière générale plus efficaces que pour les états 6s, ceci pour les trois donneurs de chlore dont nous avons donné des informations dans les tableaux 11, 12 et 13.
Les constantes de vitesse sont deux fois plus rapides pour le chlore que pour HCl et CCl4.
Pour HCl, la situation est différente du cas précédent. Si les constantes de vitesse totales sont du même ordre de grandeur que celles des états 6s, les rapports de branchement ΓXeCl sont élevés. Le résultat explique la prévision de Kannari et al.[123] quant à l’efficacité du taux de formation de XeCl* à partir de Xe(6p).
Si l’on se réfère aux courbes de potentiel de la figure 3, on constate que les courbes de potentiel V(Xe**+RX) et V(Xe++RX−) se coupent à une distance internucléaire plus grande que pour les états 6s dans une région où les interactions sont fortes[120]. Cela explique qu’après le croisement, la production de XeCl soit plus efficace que dans les états 6s[107] - [120] quelle que soit la nature du donneur de chlore, comme on peut le voir pour Cl2, HCl, CCl4 mais aussi pour les chlorofluorométhanes[127] dans les états 6p[1/2]0 et 6p[3/2]2.
Des réactions compétitives se produisent quand même. L’une d’entre elles a été observée expérimentalement et quantifiée. Il s’agit de la relaxation collisionnelle induite par HCl[128] :
Xe(6p[3/2]2) + HCl → Xe(6s[5/2]20) + HCl
avec la constante de vitesse ka où ka = 4,3.10-11 cm3s-1.
Elle ne représente que 6 % de la valeur de kQ du tableau 12 pour l’état (6p[3/2]2). Comme la proportion de la formation d’exciplexe est chiffrée à 60 %, cela indique qu’il y a d’autres processus compétitifs importants qui interviennent. Il reste à les déterminer.
Les résultats que nous venons de donner et résumés dans le tableau 12 sont relatifs à HCl(v=0). Pour les états 6p, le rôle de l’excitation vibrationnelle de HCl dans la cinétique de formation de XeCl est mal connu. Certains auteurs avancent des constantes de vitesse voisines de l’état v=0 si HCl est excité vibrationnellement. Pour cela, ils ne s’appuient sur aucune mesure directe et fondent leurs estimations sur des analogies. Là encore il apparaît nécessaire de préciser expérimentalement cette cinétique. Pour information, la constante de vitesse de formation est chiffrée pour v=1 à 5,6.10-10 cm3.s-1[121]. La même valeur est retenue pour v=2[125]. Kannari et al.[126] ne se risquent toujours pas à décliner les différents niveaux de vibration de HCl et pour v≥1, ils proposent 8,2.10-10 cm3.s-1.
États fortement excités du xénon
Les expériences menées avec Cl2 semblent indiquer que l’efficacité de formation de XeCl augmente avec l’énergie d’excitation de l’atome de xénon : la constante de formation est multiplié par trois lorsque l’on passe des états 6s aux états 7p (tableau 11).
Le taux de formation de XeCl* augmente d’un ordre de grandeur si l’on passe des états 6s aux états 6p lorsque l’on utilise CCl4 (tableau 13).
La situation est moins claire pour HCl. Comme on peut le voir sur le tableau 12, l’augmentation de kQ ne semble pas augmenter de manière significative avec l’excitation du xénon. Pour l’instant, on ne dispose pas de mesures qui vont au-delà de l’état 5d[3/2] qui est à peu près à la même énergie que les états 6p. Le taux de formation semble également très efficace à partir des états 7s[3/2][75] sans que l’on ne connaisse de valeur chiffrée. Les informations disponibles ne semblent pas confirmer l’hypothèse d’un taux de formation plus efficace de l’exciplexe au fur et à mesure que croît l’excitation du xénon. En effet, pour l’état 5d[5/2]30, on n’a observé qu’une réaction de désexcitation avec une constante de vitesse de 3,2.10–12cm3.s-1[128] :
Xe(5d[5/2]20) + HCl → Xe(6 p [3/2]2) + HCl
De la même manière, les états de Rydberg ne semblent pas produire du XeCl. En effet, les réactions observées pour Xe(31f)[129] sont les suivantes :
Xe(31f) + HCl(J) → Xe(31l) + HCl(J) (α)
Xe(31f) + HCl(J) → Xe(nl) + HCl(J-1) si J≤5 (β)
Xe(31f) + HCl(J) → Xe+ + e− + HCl(J-1) si J>5 (γ)
La constante de vitesse totale vaut kT = (11,3 ± 3,0).10–7cm3.s-1. Elle se répartit en :
kα = (5,5 ± 2,5).10–7cm3.s-1 (l-changing)
kβ = (4,8 ± 2,4).10–7cm3.s-1 (n-changing)
kγ = (0,9 ± 0,4).10–7cm3.s-1 (ionisation)
Remarquons que la réaction (γ) produit Xe+ qui est un précurseur important de XeCl comme nous allons le voir plus loin.
Conclusion à propos des réactions harpon
Il apparaît donc que les réactions harpon jouent un rôle important dans la cinétique des lasers.
Pour le laser Xe2Cl, la situation est simple avec CCl4 dont nous venons de parler. Pour le laser XeCl la cinétique relative au harponnage est plus complexe. En effet malgré sa faible proportion dans le mélange gazeux, Cl2 fournit de manière efficace de l’exciplexe par la voie du harponnage. Nous avons vu aussi que les états 6s n’interviennent dans la production de XeCl* que dans la mesure où ils donnent lieu à des collisions sur les molécules HCl excitées vibrationnellement.
La cinétique d’excitation vibrationnelle de HCl joue donc un rôle capital. Des travaux récents ont montré qu’au moins les six premiers niveaux de vibration doivent être pris en considération par bâtir un modèle satisfaisant[130] - [131] - [132] - [133]. Cette excitation vibrationnelle est produite par les électrons :
HCl(v) + e− → HCl(v’) + e− (EV)
avec la constante de vitesse K.
On a mesuré des constantes de vitesse de (EV) pour les transitions suivantes : v=0→v’=1, v=0→v’=2, v=1→ v’=2 et v=2→v’=3. Une loi empirique a été proposée[132] :
Kv→v+1 = v K0→1
Kv→v+2 = v K0→2
Les valeurs de K dépendent de la distribution d’énergie des électrons comme le montre la figure 4. Analyser plus en détail cette cinétique sort du cadre de notre travail.
Il manque également des données sur les collisions faisant intervenir HCl(v≠0) et sur celles impliquant des états du xénon fortement excités. Leur rôle dans les processus de création de XeCl* pourrait être non négligeable.
Dans les réactions harpon, le taux de formation de l’état B par rapport à celui de l’état C est compris entre 1 et 2 quel que soit l’halogénure de gaz rare[63]. On note cependant une nette augmentation de la proportion d’états B par rapport aux états C lorsque la pression croit[101]. Ce rapport est aussi fortement influencé par la nature du donneur de chlore. Il est de 1,2 pour CCl4[101] et 1,3 pour Cl2[63]. L’état d’excitation du xénon est aussi à prendre en compte. Dans le cas de Cl2, il a été observé[117] que le taux de formation de l’état B pouvait être cinquante fois supérieur à celui de C si Xe(6p[1/2]0) participe à la réaction plutôt que si ce sont des états plus fortement excités.
D’autres réactions font intervenir des collisions réactives entre espèces neutres. Elles jouent un rôle qui n’est pas négligeable, au moins pour certaines d’entre elles comme nous allons le voir maintenant.
Rôle des molécules de xénon
Nous n’avons pas trouvé dans la littérature de réactions dans lesquelles interviennent des molécules de xénon et HCl.
Lorents[75] a seulement mesuré la constante de vitesse de destruction de Xe2* par HCl sans en préciser les produits. Elle vaut : (8,2 ± 0,8).10–10cm3.s-1.
Par contre, Bibinov et Vinogradov[113] ont observé la réaction suivante avec Cl2 :
Xe2* + Cl2 → XeCl* + Cl + Xe
La formation de l’exciplexe se fait par harponnage. La constante de vitesse est estimée à 7,1.10-10 cm3.s-1[126].
Rôle de HCl excité
Castillejo et al.[134] ont observé une émission de HCl entre 200 et 240 mm qui serait due à la transition B(1Σ+) → X (1Σ+) (voir figure 5). Lorsque la pression de xénon augmente, cette émission disparaît au bénéfice d’une émission de XeCl(B). Autrement dit, XeCl(B) pourrait se former par la réaction :
HCl (B 1Σ+) + Xe (1SO) → XeCl(B) + H
La constante de vitesse est estimée à 5.10-10 cm3.s-1[135].
Une autre voie de sortie est semble-t-il compétitive à la formation de l’exciplexe à partir de la même collision ; le produit serait :
Xe+ + H + Cl + e−
et la constante de vitesse associée est 1.10-10 cm3.s-1[126].
Rôle de Cl2 excité
Cl2 se forme dans le laser par la réaction :
Cl* + HCl → Cl2* + Cl
La constante de vitesse est 1.10-10 cm3.s-1[126]. La production de l’exciplexe se fait alors par la réaction :
Xe + Cl2*(1Σu+) → XeCl*+ Cl
avec la constante de vitesse ku
Les valeurs de ku sont données dans le tableau 14. On constate que la valeur de Zuev et al.[136] est statistiquement éloigné des autres bien qu’il s’agisse d’une mesure récente. Sans en tenir compte, la valeur moyenne est ku = 2,6.10-10 cm3.s-1.
Tableau 14 : Valeurs de ku en cm3s-1 trouvées dans la littérature.
Une réaction analogue se produit pour l’état Cl2* (D’ 3π2g)[113]. On n’a pas d’autre renseignement à ce sujet.
Réactions à trois corps
Elles se produisent essentiellement dans les mélanges ternaires. Elles sont du type :
Xe** + Cl2 + M → XeCl* + Cl + M avec la constante de vitesse kc
Les constantes de vitesse kc sont données dans le tableau 15. On constate que les processus où M=Ar sont négligeables.
État du xénon Xe** M = Xe × 10-28 M = Ar × 10-28 (6p[1/2]0) (3,5 ± 0,5) < 0,5 (6p[3/2]2) (1,4 ± 0,5) < 0,1 (6p[5/2]2) (1,8 ± 0,5) < 0,1
Tableau 15 : Valeurs de kc en cm6s-1 tirées de Bruce et al.[117].
Pour l’hélium, on dispose de deux réactions :
Xe* + Cl + He → XeCl* + He
Xe** + Cl + He → XeCl* + He
Les constantes de vitesse valent respectivement 10-27 cm6.s-1 et 3.10-27 cm6.s-1[139].
Il existe aussi des données où les atomes de xénon sont à l’état fondamental :
Xe + Cl + M → XeCl (X) + M où M = Ne ou Xe
Dans les deux cas, la constante de vitesse vaut : 1,2.10-33 cm6.s-1[140].
Autres réactions
Le chlore Cl2 formé dans le mélange gazeux peut induire les réactions suivantes :
Xe + Cl2 → XeCl2
Xe* + Cl2 + Xe → Xe+ + Cl2− + Xe → (XeCl2)* + Xe[141]
Comme la température de sublimation de XeCl2 vaut ts= 80 °C, cette molécule est formée dans les conditions de température ambiante, à l’état solide dans le mélange gazeux. Ce qui provoque un phénomène parasite dit « laser snow »[142].
Certains auteurs ont proposé d’augmenter la température afin de sublimer XeCl2. Il devient alors réactif et participe activement à la formation de XeCl* :
XeCl2* → XeCl* + Cl
Xe* + XeCl2 → 2 XeCl*
L’augmentation de température a donc un double intérêt : éliminer un phénomène parasite et augmenter la production de XeCl. Toutefois, l’élévation ne doit pas être trop importante afin de ne pas dissocier XeCl2, ce qui éliminerait les réactions précédentes.
Dans les mélanges ternaires des exciplexes du type RgCl peuvent être formés. Ils peuvent conduire à la création de XeCl* par des réactions dites de déplacement. Elles ont été observées lorsque Rg = Ar ou Kr[140] - [143] :
RgCl* + Xe → XeCl* + Rg
avec la constante de vitesse kd où kd=1,5.10-10 cm3.s-1 pour Rg = Ar
À l’inverse, la formation de RgCl consomme du chlore qui fait baisser le taux de production de XeCl. La qualité du laser peut en pâtir comme c’est le cas avec le krypton[144].
Nous nous limiterons à cet examen des réactions de formation de XeCl* hors recombinaison ionique. Cette seconde voie, de loin la plus importante, va être examinée maintenant.
Les recombinaisons d’ions
Selon plusieurs auteurs[120] - [145] - [146] les réactions à deux corps (Xe+ + Cl−, Xe2+ + Cl− et RgXe+ + Cl−) n’interviennent pas. Les réactions ternaires sont du type :
Xe+ + Cl− + Rg → XeCl* + Rg (3)
Xe2+ + Cl− + Rg → XeCl* + Rg + Xe (4)
RgXe+ + Cl− + Rg → XeCl* + 2 Rg (5)
Les ions contenant du xénon sont formés directement dans la décharge ou par une succession de réactions faisant intervenir Rg+, Rg2+ et d’autres espèces ioniques ou excitées. La figure 1 montre un exemple où Rg=Ne et la figure 6 où Rg=He. Un examen approfondi de ces réactions sort du cadre de cet article. Le lecteur pourra en trouver des exemples dans des publications traitant de la modélisation du milieu laser[121] - [124] - [147] - [135] - [148] - [149].
Les ions Cl− se forment essentiellement par attachement dissociatif d’un électron sur HCl[38] :
HCl(v) + e− → H + Cl− (AD)
Là encore les constantes de vitesse de (AD) dépendent de la distribution d’énergie des électrons comme l’illustre la figure 4.
Le troisième corps Rg est chimiquement passif. Il ne sert qu’à la stabilisation de la réaction[150]. De ce fait, les auteurs ne prennent en considération que le taux de recombinaison des ions positifs et négatifs. Celui-ci varie de façon significative avec la pression totale du mélange gazeux, la nature du gaz tampon et la température.
Les réactions (3) et (4) ont été mises en évidence expérimentalement pour tous les gaz rares. Les figures 7 et 8 montrent l’influence du gaz tampon et de la pression sur le taux de recombinaison de ces réactions lorsque l’on utilise l’hélium puis le néon comme gaz tampon. Ce taux de recombinaison est du même ordre de grandeur dans les deux cas. Il est de quelque 10-6 cm3.s-1. L’influence de la température ne semble avoir été étudié que dans le cas du néon. La figure 9 synthétise l’ensemble des travaux effectués. Le taux de recombinaison α3 dans la réaction (3) est maximal à 180K pour une pression totale de 294,2 kPa[151]. α3 vaut alors 4,2.10-6 cm3.s-1.
L’analyse la plus fine de la réaction (4) a été effectuée par Bates et Morgan[152]. Ils font remarquer que les méthodes Monte-Carlo, l’équation de Flannery et la théorie de Langevin ne donnent de bons résultats que pour des pressions supérieures à 1 atm. Notons que c’est toujours le cas dans les lasers. La théorie « tidal » qu’ils proposent est en très bon accord avec les mesures expérimentales de Mezyk et al.[145] comme on peut le voir sur la figure 10. On note que le taux de recombinaison α4 pour la réaction (4) est du même ordre de grandeur que α3.
La réaction (5) n’a été observée que lorsque Rg = Ne ou Rg = Ar. Pour cette réaction, l’évolution du taux de recombinaison α5 avec la pression de néon est montrée sur la figure 6. L’influence de la température a été étudié par Imada et al.[153] pour une pression totale fixée à 294 kPa. La valeur maximale de α5 est obtenue à 120 K et α5 = 7,5.10-6 cm3.s-1.
Dans le cas de l’argon, nous n’avons que deux estimations à température ambiante. Pour une pression de 2 atm, α5 = 2.10-6 cm3.s-1[154] et pour une pression de 1 atm, α5 vaut 1.10-6 cm3.s-1[70].
La réaction (5) ne favorise pas le passage par un complexe transitoire RgXeCl*[62]. La réaction suivante a donc un rôle mineur :
RgXe+ + Cl− + Rg → RgXeCl* + Rg → XeCl* + 2 Rg
Au contraire, la principale voie de formation se fait par :
RgXe+ + Cl− + Rg → 2 Rg + Xe+ + Cl− → XeCl* + 2Rg
Les calculs de Kannari et al.[147] chiffrent la contribution de chacune des trois réactions de recombinaison et des réactions de harponnage pour trois types de mélanges. Les résultats sont rassemblés dans le tableau 16. On constate que c’est la réaction (3) qui fournit l’essentiel des exciplexes et d’une manière générale les réactions de harponnage jouent un rôle secondaire. Dans l’hélium par contre, les réactions de harponnage constituent pour certains auteurs une contribution de 10 à 15 % à la formation de XeCl*[149] - [155]. D’autres ne l’estiment qu’à 1 % des voies ioniques[130]. Ces conclusions théoriques sont confirmées par la voie expérimentale pour l’ensemble des gaz tampons et pour d’autres donneurs de chlore[149] - [156]. Les réactions « harpoon » sont quand même importantes malgré leur faible contribution. En effet, ce sont elles qui se déroulent en premier sitôt après l’excitation. Les recombinaisons d’ions ne débutent que 20ns plus tard pour alors fournir l’essentiel des exciplexes[149].
Réaction Xe/HCl Ar/Xe/HCl Ne/Xe/HCl Xe+ + Cl− 83,1 % 81,5 % 69,6 % Xe2+ + Cl− 11,9 8,2 9,5 MXe+ + Cl− 6,3 11,1 Xe** + HCl 2,5 1,4 1,4 Xe* + HCl(v) 2,5 2,6 2,6 Autres 5,8
Tableau 16 : Contributions en pourcentages des réactions de formation de XeCl*. Les paramètres d’excitation sont : taux d’excitation de l’ordre de 3 MW/cm3 et la largeur du pulse de pompage est de 55 ns.
Remarquons dans le tableau 16, la ligne « autres » qui indique 5,8 % dans la colonne du néon. Cela montre que d’autres voies sont ouvertes. Examinons maintenant, les autres réactions de recombinaison qui ont été signalées dans la littérature.
Les ions Xe3+ se forment dans les mélanges gazeux utilisés dans les lasers. Ces ions se recombinent avec Cl− pour donner XeCl. Toutefois, cette réaction n’a semble-t-il qu’une faible contribution dans la cinétique du laser[157].
Des travaux récents montrent que les ions Xe+* réagissent avec Cl− pour donner XeCl*[21] - [158]. Ainsi, Alekhin et al.[158] ont formé du XeCl* à partir de vapeurs de NaCl. XeCl* est produit dans les états vibrationnels les plus bas (v≤20) à partir d’un ion Xe+* fortement excité dans une réaction à deux corps. La constante de formation est estimée entre 2.10-10 et 1.10-9 cm3.s-1. Une réaction analogue est proposée à partir de HCl[21]. Cette conclusion repose sur l’intervention des états responsables du troisième continuum du xénon qui ne peuvent être des ions Xe2+ car ils ne produisent pas de XeCl*[151] - [153]. Au contraire, la participation d’ions Xe+* est compatible avec les observations faites par ailleurs. Plusieurs auteurs[149] - [155] - [159] confirment la présence de l’ion Xe+*(6s 4P3/2) dans le mélange laser qu’ils étudient. Leur concentration serait mille fois supérieure à celle de Xe* responsable des réactions harpon[130]. D’autre part, la dépendance temporelle des concentrations de ces ions et de celles de XeCl* et Cl−, n’est pas incompatible avec une voie de formation de l’exciplexe à partir de Xe+*. En effet, le début de décroissance de Xe+* et de Cl− correspond à une accélération croissante du taux de formation de XeCl*. Cependant, il reste à approfondir la connaissance de ces phénomènes. Une autre question importante, est la répartition entre les différents états B, C et D après la formation de XeCl. Nous avons vu que pour les réactions « harpon », la répartition se fait entre les états B et C dans des proportions très variables suivant les conditions expérimentales.
Pour la voie ionique, une première estimation a été faite par Tisone et Hoffman[160] qui proposent 76 % d’états B et 24 % d’états C. Les gaz tampons sont successivement Ne, Ar, et Kr. Ohwa et Kushner[161] annoncent des valeurs similaires : 77 % d’états B et 23 % d’états C. Ils utilisent un mélange quaternaire qui contient en plus du gaz tampon (du néon) de l’hydrogène H2.
Une étude récente et plus détaillée a été menée par Tsuji et al.[146] dans un mélange qui utilise l’hélium comme gaz tampon. Ils trouvent que :
- les états D sont surtout formés à partir de l’ion Xe+(2P1/2) ;
- les états B et C sont exclusivement produits à partir de l’ion Xe+(2P3/2) dans les proportions suivantes : (62,6 %) d’états B pour (38,4 %) d’états C. Le taux de production de XeCl* est de 98 %[162]. Il y a donc peu de réactions compétitives.
Dans l’expérience, le nombre d’états Xe+(2P1/2) et Xe+(2P3/2) est le même. De plus, les constantes de vitesse des réactions (3) relatives à ces deux états du xénon sont voisines. Pourtant, dans ces conditions, le nombre d’états D formés est très faible par rapport au nombre d’états B et C. Le taux de formation de XeCl(D) par rapport à celui de XeCl(B, C) est estimé à 0,033±0,006. La prédissociation plus rapide de [Xe+(2P1/2)Cl−]* par rapport à celle de [Xe+(2P3/2)Cl−]* est responsable de cette situation.
Spectres d’émission
Les spectres analogues à ceux de la figure 11 sont observés par la quasi-totalité des auteurs qui étudient les mélanges à base de xénon et d’un donneur de chlore. On présente ici quelques-uns de ces résultats, parmi les plus significatifs.
Deux études théoriques permettent d’identifier les émissions des spectres[48] - [54]. On y prévoit cinq transitions d’intensité élevée qui correspondent à ΔΩ = 0, c’est-à-dire polarisées parallèlement à l’axe internucléaire. Les états de départ sont toujours de type ionique et les états d’arrivée de type covalent. Les caractéristiques des émissions sont consignées dans le tableau 17.
Transition Expérience Théorie Théorie Théorie Longueur d’onde observée (nm) Longueur d’onde d’émission calculée (nm) Moment de transition (s) Probabilité d’émission (s-1)x 107 B → X 308[70] 295[54] ; 282[48] 2,76[54] ; 2,85[48] 9,3[54] ; 11,4[48] D → X 235,5[58] 224[54] ; 216[48] 1,94[54] ; 2,09[48] 10[54] ; 14[48] C → A3/2 345[70] 330[54] ; 306[48] ; 355[92] 0,96[54] ; 0,98[48] 0,81[54] ; 1,05[48] B → A1/2 345[70] 324[54] ; 307[48] 0,87[54] ; 0,88[48] 0,6[54] ; 0,84[48] D → A1/2 Non observée 242[54] ; 233[48] 0,50[54] ; 0,49[48] 0,56[54] ; 0,59[48]
Tableau 17 : Émissions de XeCl*.
On y voit que les transitions UV les plus probables sont B→X et D→X. Elles sont de nature Σ→Σ. Les autres transitions B→A, C→A et D→A sont de nature Π→Π et sont beaucoup moins probables[78].
D’autres transitions théoriquement plus faibles n’ont pas encore donné lieu à une observation à une exception près. En effet, Hay et Dunning[54] prévoient quatre transitions polarisées perpendiculairement à l’axe internucléaire, c’est-à-dire avec ΔΩ = ±1. Seuls Ewing et Brau[94] indiquent une émission centrée à 425 nm qu’ils attribuent à une transition 2Σ→2Π. Enfin, Krauss[78] signale la possibilité d’une émission de type D→B dont le moment de transition est lui aussi très faible. Le tableau 6 permet de la situer à 931nm.
Intéressons-nous maintenant aux principales émissions observées et signalées dans le tableau 17.
La bande B→X est observée à 308 nm (figure 11) alors que la prévision théorique est nettement plus faible. C’est la plus étroite des émissions ; l’état d’arrivée présente en effet un puits de potentiel, même s’il est un peu profond. Comme dans tous les halogénures de gaz rares, c’est l’émission qui a le plus fort moment de transition. C’est pourquoi, c’est l’émission qui est utilisée dans les lasers XeCl[6].
Expérimentalement, les bandes (C→A) et (B→A) se chevauchent[64] et donnent lieu à un continuum centré à 345 nm qui est souvent de faible amplitude comme on peut le voir sur la figure 11. La largeur des émissions tient à ce que la transition s’effectue vers un état fortement répulsif. Koltz et al.[55] situent ce continuum entre 312 et 460 nm. La faible intensité observée s’explique par la faiblesse des probabilités de transition des deux émissions en regard de celle de B→X et par le peu d’états C formés en regard de l’état B comme nous l’avons déjà vu. Par ailleurs, d’autres auteurs signalent un phénomène d’absorption de la molécule Xe2Cl à cette longueur d’onde[163]. Il est aussi intéressant de chiffrer la contribution de la transition (B→A) dans l’émission centrée à 345 nm. D’après Kannari et al.[147], la voie principale de formation des états B et C est la réaction (3). Tsuji et al.[146] ont estimé la proportion d’états B et C ainsi formés : elle est de 38 % pour les états C et de 62 % pour les états B. Compte tenu des valeurs des probabilités de transition (valeur théorique de IB→A/IB→X = 0,07 ; valeur expérimentale 0,05[55], la contribution de l’émission (B→A) est d’environ 10 %. Plusieurs auteurs[8] - [64] - [164] pensent qu’il est possible de développer un laser basé sur l’émission à 345 nm, en particulier à forte pression (de l’ordre de 10 atm) lorsque les états B et C sont thermalisés. Cependant, aucun résultat concret n’est signalé à ce jour.
La transition (D→X) centrée à 235,5 nm n’est pas observée de manière systématique. La bande correspondante apparaît faiblement dans le cas de la figure 12. Sa largeur optique est analogue à l’émission (B→X) car elle aboutit sur le même état X qui est faiblement lié[58]. Par contre, l’intensité relative des émissions (B→X) et (D→X) varie considérablement d’un auteur à l’autre : ID→X/IB→X = 1/3 pour Shuker[58], 1/25 à 1/50 pour Sur et al.[86] et 0,14 pour Taylor et al.[165]. Ces derniers notent que le rapport est indépendant de la pression. Ainsi, il paraît peu probable qu’un laser soit développé sur cette transition comme le croyait Shuker[58].
Quant à l’émission D→A, elle n’est pas signalée dans les spectres. Indiquons toutefois qu’Hassal et Ballik[105] observent une bande à 246 nm de très faible intensité (figure 12) sans l’attribuer à la transition considérée, ce qui serait pourtant possible.
En conclusion, nous pouvons dire que les émissions dont l’état D est l’état de départ, jouent un rôle négligeable dans la spectroscopie de XeCl. Si les absences de D→A comme de D→B peuvent s’expliquer par la faible probabilité de transition associée[48] - [54] - [78] il n’en est pas de même pour D→X. En effet, si l’on se réfère au tableau 17, l’émission D→X devrait être légèrement plus intense que B→X. Dans ce cas, l’explication peut être la faible production d’états D que ce soit par la voie ionique[146] ou par harponnage à partir des états Xe(3P)[102]. Si nous nous référons encore au fait que la voie principale de formation de XeCl* est la réaction (3) et compte tenu des résultats de Tsuji et al.[146], on peut estimer que le rapport du nombre d’états B à celui de l’état D vaut 0,053. Compte tenu des observations et des probabilités de transition du tableau 17, on peut penser que les états D se désexcitent exclusivement vers l’état X. En prenant en compte les probabilités de transition du tableau 17, on arrive à ID→X/IB→X≈6,2 %. Ce qui est de l’ordre de grandeur des observations de Sur et al.[86] et pas très éloigné de Taylor et al.[165].
Toutes ces émissions sont plus ou moins dégradées du côté des courtes longueurs d’onde comme le montre le spectre d’émission de la bande (B→X) sur la figure 13. Un phénomène d’oscillations analogue et aux mêmes longueurs d’onde a été observé dans des spectres d’absorption[57]. Par ailleurs, l’émission (D→X) a la même structure de bande que (B→X)[86].
La largeur et la nature oscillatoire de ces bandes sont liées à l’existence de transitions à partir de niveaux vibrationnels élevés de l’état radiatif[55] - [79] - [97]. L’excitation vibrationnelle provient de l’énergie restante après la formation de l’exciplexe. Cette énergie dépend à la fois de l’état de l’atome ou de l’ion de xénon impliqué dans la réaction et du donneur d’halogène[63] - [79] - [120]. Pour l’émission à 345 nm, les transitions à haut niveau vibrationnel sont plus décalées vers le rouge pour C→A3/2 que pour B→A1/2 car la barrière répulsive de A3/2 est plus pentue et plus proche de l’état supérieur de l’émission qu'A1/2[79].
La forme oscillatoire des spectres tend à disparaître avec l’augmentation de la pression pour ne plus présenter que des pics issus des niveaux v≤2 au-delà de 1atm. Ce qui indique que la relaxation vibrationnelle dépeuple de façon efficace les niveaux vibrationnels les plus hauts[13] - [97]. D’autre part, la disparition des niveaux élevés est plus rapide pour l’état B que pour l’état C car ce dernier a une durée de vie beaucoup plus longue[79]. On peut donc penser que la relaxation vibrationnelle des états B et C va jouer un rôle important dans la cinétique du laser XeCl.
Si la pression augmente la situation se complique encore. Au-delà de 5atm, un élargissement de ces bandes se manifeste. Il se pourrait qu’il s’agisse d’un élargissement collisionnel induit sur des raies ou sur l’ensemble de la structure rotationnelle[166].
Les effets isotopiques sont négligeables pour le xénon mais ils sont sensibles pour le chlore. Les bandes vibrationnelles associées à l’isotope le plus lourd 37Cl, sont légèrement déplacées vers les plus grandes longueurs d’onde. Par exemple, l’écart peut atteindre 1,51Å pour la bande 4-0 de B→X[60].
Durées de vie radiative des espèces excitées
Les valeurs obtenues pour les états B, C et D sont rassemblés dans le tableau 18 pour ce qui concerne le niveau vibrationnel v=0. Ce sont les états B et C qui ont donné lieu au plus de déterminations.
État B : τB État C : τC État D : τD Méthode Référence 11,1 ± 0,2 130,5 ± 1,5 Expérimentale (gaz) Asselman et al.[23] 27 ± 3 53 ± 6 Expérimentale (gaz) Grieneisen et al.[72] 10,1 123 9,5 Théorique Hay et Dunning[54] 11,1 ± 0,2 131 ± 10 Expérimentale (gaz) Inoue et al.[66] 135 Expérimental (gaz) Yu et al.[74] 8,2 95 6,9 Théorique Adams et Chabalowski[48] 11 Expérimentale (solide) Le Calvé et al.[96] 133,5 ± 4,5 Expérimentale (solide) Fajardo et Apkarian[77] 120 ± 9 Expérimentale (solide) Böhling et al.[81] 17 Expérimentale (gaz) Fisher[167]
Tableau 18 : Durées de vie (en ns) des états de XeCl*.
Pour l’état B, deux valeurs sont statistiquement éloignées des autres[72] - [167]. Elles correspondent aux mesures les plus anciennes. Sans en tenir compte, on obtient comme intervalle de confiance en ns : 8<τB<12,3.
Pour l’état C, la dispersion est plus importante. La détermination de Grieneisen et al.[72] est encore statistiquement éloignée des autres ainsi que les deux valeurs théoriques[54] - [48] et une mesure obtenue à l’état solide[81]. Sans en tenir compte, l’intervalle de confiance est en ns : 129,1<τC<135,9.
Avec les valeurs moyennes, le rapport τB/τC est de 0,0764. Il est à comparer avec une mesure directe qui vaut : 0,087 ± 0,009[69]. L’accord est convenable. La connaissance précise de ce rapport est importante car il joue un rôle dans la relaxation vibrationnelle des états B et C comme nous l’avons vu au paragraphe précédent.
Par ailleurs, une étude systématique des durées de vie de plusieurs niveaux vibrationnels (v≤136) des états B et C a été menée à bien[168]. Les résultats sont consignés dans le tableau 19.
Niveau de vibration Énergie (cm-1) ; État C Durée de vie (ns) ; État C Énergie (cm-1) ; État B Durée de vie (ns) ; État B 0 139,42 120,0 369,42 11,0 4 876,08 127,6 1 136,05 11,08 8 1 590,86 136,4 1 882,33 11,88 12 2 284,25 137,2 2 608,63 12,29 16 2 956,77 142,8 3 315,38 12,64 20 3 608,94 146,9 4 002,98 12,53 24 4 241,29 152,3 4 671,84 12,35 28 4 854,33 174,1 5 322,39 13,43 32 5 448,6 182,1 5 955,05 14,10 36 6 024,61 195,3 6 570,25 14,5 40 6 582,89 195,5 7 168,42 14,84 44 7 123,96 210,3 7 750,00 16,12 48 7 648,33 224,6 8 315,41 16,38 52 8 156,52 230,6 8 865,10 17,25 56 8 649,03 245,0 9 399,49 18,69 60 9 126,35 256,4 9 919,03 19,33 64 9 588,98 265,0 10 424,17 20,15 68 10 037,4 275,2 10 915,27 21,35 72 10 472,1 279,1 11 392,77 22,42 76 10 883,4 270,2 11 897,07 23,88 80 11 302,0 296,2 12 308,67 24,78 84 11 698,1 298,2 12 747,97 26,04 88 12 082,3 308,3 13 175,27 27,52 92 12 454,9 318,1 13 390,97 28,98 96 12 815,3 325,6 13 994,47 30,21 100 13 167 337,7 14 389,17 31,77 104 13 507,3 343,3 14 772,37 33,21 108 13 837,6 349,1 15 145,174 35,14 112 14 158,1 352,8 15 508,67 37,16 116 14 469,3 357,9 15 862,27 39,03 120 14 771,5 375,1 16 206,67 40,91 124 15 065 398,5 16541,97 128 15 627,1 433,7 17 186,47 136 15 896,2 438,5 17 496,07
Tableau 19 : Durées de vie des niveaux vibrationnels des états B et C de XeCl[168].
On constatera que les durées de vie augmentent d’un facteur 4 lorsque v passe de 0 à 100. Une représentation graphique extrapolée des données relatives à l’état B est présentée sur la figure 14.
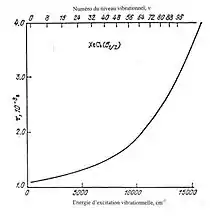
Pour l’état D, il n’y a que trois déterminations relativement proches les unes des autres. D’autre part, à l’état gazeux, Shuker[58] a signalé que l’émission D→X avait une dépendance temporelle similaire à B→X, ce qui va dans le sens des grandeurs précédentes puisque la durée de vie de l’état B est de l’ordre de 10ns. Cependant, d’autres mesures sont nécessaires pour avoir une valeur précise de τD.
Voie collisionnelle
Dans un souci de clarté, nous présenterons tout d’abord les influences du xénon et de HCl. Ensuite, nous étudierons le rôle des divers gaz tampons puis celui des donneurs de chlore.
Dans les mélanges Xe/HCl
Les seuls processus de destruction des états B et C de XeCl, autres que le processus radiatif, qui ont été mis en évidence sont :
XeCl* + HCl → produits autres que XeCl (6)
avec la constante de vitesse kH
XeCl* + Xe → produits autres que XeCl (7)
avec la constante de vitesse kX
XeCl* + 2 Xe → produits autres que XeCl et Xe2Cl ou → Xe2Cl* + Xe (8)
avec la constante de vitesse kDX
XeCl* + Xe + HCl → produits autres que XeCl (9)
avec la constante de vitesse kM
XeCl* + e− → Xe + Cl + e− (10)
avec la constante de vitesse ke
Nous n’avons trouvé aucun résultat concernant l’état D.
Les valeurs obtenues pour les états B et C sont réunies dans le tableau 20. Notons que les auteurs supposent que les taux de réaction sont identiques pour les deux états.
Réf kH kX kDX kM ke Finn et al.[169] 1,4 × 10-9 (± 40 %) 3,2 × 10-11 (± 35 %) Inoue et al.[66] (6,3 ± 0,5) × 10-10 (2,3 ± 0,3) × 10-11 Turner et al.[135] 4 × 10-8 Le Calvé et al.[76] 0,4 × 10-11 1,3 × 10-30 Lorents[75] (7,3 ± 0,1) × 10-10 < 4 × 10-12 (1,53 ± 0,1) × 10-30 Quiñones et al.[67] (5,0+3,0-2,0) × 10-12 (13,0 ± 4,0) × 10-31 Tang et al.[170] 7,3 × 10-31 Qihong[171] 1,16 × 10-7 Tysone et Hoffman[160] 1,7 × 10-9 4 × 10-31 1,2 × 10-7 Yu et al.[172] (7,3 ± 0,1) × 10-10 Zuev et al.[136] 1,5 × 10-30 Fisher[167] 7,7 × 10-10 2,1 × 10-12 1 × 10-30 Rives et al.[22] (3,8 ± 2,3) × 10-10 (4 ± 19) × 10-13 (1,0 ± 0,4) × 10-30 (4,6 ± 2,1) × 10-29 Hokazono et al.[173] 1,5 × 10-31 Johnson et al.[140] 5 × 10-31 2 × 10-8 Levin et al.[121] 3 × 10-7 Maeda et al.[174] 3 × 10-8 Baginskii et al.[175] 2 × 10-7 Ishihara et Lin[124] 1 × 10-7
Tableau 20 : Constantes de vitesse de disparition de XeCl(B, C) en cm3s-1pour ke, kH et kX et en cm6s-1 pour kDX et kM.
On constate que la réaction (9) n’a été observée qu’une seule fois et ce récemment[22]. Il n’y a donc aucun élément de comparaison disponible.
Par contre, les autres réactions ont été observées et quantifiées à de nombreuses reprises.
Pour kH, trois mesures sont statistiquement éloignées des autres[22] - [160] - [169]. Les deux dernières, anciennes, sont très largement supérieures aux autres. La première, récente, est la seule expérience à mettre en évidence le processus (9) qui a toujours été négligé auparavant. Si l’on fait de même avec les mesures de Rives et al.[22], kH est alors multiplié par 2, ce qui la ramène au même niveau que les autres valeurs. Si l’on prend en compte la réaction (9), force est de constater que l’ensemble des valeurs de kH doit être revu à la baisse sauf Rives et al.[22]. Nous ne pouvons donc pas donner d’intervalle de confiance dans ces conditions.
Pour kX, l’analyse statistique est très délicate car il y a une très forte dispersion des valeurs doublée d’incertitudes absolues importantes. Lorents[75] ne fournit qu’une limite supérieure. Les résultats de Rives et al.[22] amènent à se poser la question de savoir si ce processus est réellement chiffrable compte tenu de la faible valeur de la constante de vitesse. Statistiquement, kX ne devrait pas dépasser 6,12.10-12 cm3s-1. Deux mesures sont alors trop fortes[66] - [169]. La dernière, ancienne, a déjà fourni une valeur erronée de kH. Pour la première, indiquons que la même équipe a revu fortement à la baisse sa mesure six ans plus tard[67].
La réaction (8) qui ne conduit pas à la production de Xe2Cl* a une importance négligeable[67] - [116]. Les mesures de kDX sont très dispersées et l’intervalle de confiance n’inclut que trois valeurs[22] - [167] - [170]. Si deux des mesures exclues sont des estimations douteuses[140] - [173], d’autres correspondent à des mesures directes[75] - [76] - [67] - [136] - [160] qui ont par ailleurs fourni de bons résultats. Une grande incertitude règne sur kDX. Bornons–nous à donner la valeur moyenne obtenue qui est représentative de l’ensemble des résultats, soit 9,1.10-31 cm6s-1.
Une très forte dispersion est encore la conclusion de l’examen des valeurs de ke. Seules quatre valeurs sont statistiquement proches[124] - [135] - [160] - [171]. Contentons-nous d’indiquer la valeur moyenne qui est de 9,6.10-8 cm3s-1 quantité relativement proche de la seule mesure directe[171].
Lou[176] propose d’autres produits pour la réaction (10) :
XeCl* + e− → Xe+ + Cl− (ke1 = 1,8.10-7 cm3s-1) ou → Xe* + Cl + e− (ke2 = 1,2.10-7 cm3s-1)
Des différences ont été constatées pour des réactions de type (6) lorsque l’on tient compte des niveaux vibrationnels des partenaires de la collision :
XeCl*(v=0) + HCl(v=1) → Xe + HCl + Cl + Cl (6a) avec la constante de vitesse kHa
XeCl*(v=0) + HCl(v=2) → Xe + HCl + Cl + Cl (6b) avec la constante de vitesse kHb
XeCl(B,C;v≠0) + HCl(v=0) → produits autres que XeCl (6c) avec la constante de vitesse kHc
Les valeurs des constantes de vitesse sont rassemblées dans le tableau 21. Elles sont très dispersées et ne correspondent à aucune mesure directe. Il s’agit uniquement d’estimations fondées sur des analogies.
Tableau 21 : Valeurs de kHa, kHb, kHc en cm3s-1.
Des réactions analogues à (6) et (7) se manifestent lorsque XeCl est dans l’état fondamental X(v=0). Ces phénomènes influent dans les performances des lasers et sont donc importants. Les constantes de vitesse sont rassemblées dans le tableau 22. Elles ne varient pas avec le niveau de vibration des molécules en collision. Notons qu’il n’y a qu’une seule mesure directe[36], les autres sont des estimations.
Tableau 22 : Constantes de vitesse en cm3s-1 de disparition binaire relatives à XeCl(X, v = 0) et un autre partenaire Xe, HCl et électron.
Rôle du gaz tampon
L’addition d’un troisième gaz en quantité importante influe aussi sur la cinétique de disparition de XeCl(B,C). Elle induit des réactions qui sont du même type que celles produites par le xénon :
Collision double (11) : XeCl(B,C) + Rg → Xe + Cl + Rg avec la constante de vitesse k11
Collision triple (12) : XeCl(B,C) + 2 Rg → Xe + Cl + 2 Rg avec la constante de vitesse k12
Collision triple mixte (13) : XeCl(B,C) + Xe + Rg → 2 Xe+ Cl + Rg avec la constante de vitesse k13
Les constantes de vitesse de ces trois processus sont regroupées dans les tableaux 23, 24 et 25.
Tableau 23 : Valeurs de k11 en cm3s-1 pour les différents gaz rares.
Les réactions (11) et (13) sont toujours importantes alors que (12) a une contribution négligeable. On ne peut que constater la grande dispersion des résultats ; des différences de plusieurs ordres de grandeur sont parfois observées. Indiquons que seules quatre références[75] - [67] - [169] - [180] sont des travaux expérimentaux produisant des mesures directes des taux de réaction. Les autres ne sont que des estimations faites par des équipes de modélisation. Elles sont basées sur des analogies. Elles ne sont données qu’à titre indicatif. Dans un modèle cinétique, il vaut mieux leur préférer les mesures directes. On notera enfin le manque de renseignements relatif au cas du krypton.
Tableau 24 : Valeurs de k12 en cm6s-1 pour les différents gaz rares.
Des réactions compétitives se manifestent pour l’ensemble de ces réactions. Examinons-les.
Réf He Ne Ar Kr Glass et al.[180]. (3,8 ± 0,2) × 10-30 Quiñones et al.[67] (2,4 ± 0,5) × 10-31 (7,4 ± 1,5) × 10-31 (8,9 ± 1,9) × 10-31 (9,9 ± 1,9) × 10-31 Lorents[75] (1,01 ± 0,05) × 10-30 Hokazono et al.[173] 1,5 × 10-32 1,5 × 10-31 Ohwa et Kushner[161] 5 × 10-32 Turner et Smith[135] 1 × 10-31 Mizunami et al.[181] 1,5 × 10-31 Ishihara et Lin[124] 2 × 10-31
Tableau 25 : Valeurs de k13 en cm6s-1 pour les différents gaz rares.
Des réactions de déplacement sont compétitives à (11). Dans ce cas, les produits sont RgCl(B). On n’en a pour l’instant observé que dans le cas où Rg = Kr[143] :
XeCl* + Kr → KrCl + Xe
La constante de vitesse vaut : 0,7.10-9cm3s-1[144]. Cette réaction est donc plus efficace que le quenching. Elle joue un rôle important dans la cinétique des lasers. Elle est aussi rapide que les processus de création de XeCl* par harponnage. Si l’on se réfère au tableau 20, il s’agit d’une des voies principales de destruction de l’exciplexe.
Pour Brashears et al.[182], il est aussi possible d’obtenir comme produit le complexe triatomique RgXeCl*. C’est donc une réaction compétitive à la collision qui produit des atomes dissociés. On a observé les émissions de KrXeCl à 370 nm[182], ArXeCl à 326 nm[183] et NeXeCl à 434 nm[96] ce qui indique bien la formation de ces complexes. On n’a mesuré la constante de vitesse que pour Rg = Kr ; elle vaut 9.10-33 cm6s-1[67].
Toutefois, la création de ArXeCl semble se faire préférentiellement par une réaction compétitive à (13) :
Xe* + Ar + Xe → ArXeCl*
La constante de vitesse vaut 4.10-30 cm6s-1[11]. Elle est donc du même ordre de grandeur que (13).
Cependant, c’est la formation du trimère Xe2Cl* qui est la réaction compétitive à (13) la plus fréquente. Ces réactions seront étudiées au chapitre suivant.
Par ailleurs, dans le cas de l’hélium, Baginskii et al.[179] prévoient une voie de sortie avec Xe2* + Cl + He dont la constante de vitesse est 1,5.10-31 cm6s-1.
Une réaction analogue à (11) se manifeste aussi pour XeCl à l’état fondamental. Les constantes de vitesse sont rassemblées sur le tableau 26. On constate une fois encore la grande dispersion des mesures et l’absence de données relatives au krypton. Signalons la présence que d’une seule mesure directe[36]. Les autres ne sont que des estimations plus ou moins fondées. Parmi elles, l’une[184] est statistiquement éloignée des autres. Notons que pour le néon, on a aussi estimé la constante de vitesse pour XeCl(X, v = 1), elle vaut 1.10-11cm3s-1[161].
Tableau 26 : Constantes de vitesse en cm3s-1 de disparition par collisions binaires relatives à XeCl(X, v=0) pour divers gaz tampons.
Influence d’autres donneurs de chlore et autres réactions
La principale réaction que nous avons relevée dans la littérature sont des réactions analogues à la réaction (6) :
XeCl* + RCl → produits autres que XeCl (14) avec la constante de vitesse kR
Les valeurs des constantes de vitesse par RCl = Cl2 ou CCl4 sont rassemblées dans le tableau 27. On y constate que les trois donneurs de chlore étudiés (HCl, Cl2 et CCl4) ont des taux de quenching du même ordre de grandeur.
Tableau 27 : Constantes de vitesse en cm3s-1 relatives aux réactions (14) pour XeCl (B, C ; v’ = 0,1).
L’ensemble des mesures du tableau 27 ont été obtenues par la voie expérimentale. Pour le chlore, seule une valeur est statistiquement éloignée des autres[66]. Pourtant il s’agit d’une mesure récente. Notons que l’écart absolu n’est très fort avec les autres déterminations. Bornons-nous à donner une valeur moyenne de kR pour le chlore : 5.10-10 cm3s-1. Elle est très proche d’une mesure relative au CCl4.
Dans le cas du chlore, Grieneisen et al.[72] signalent deux valeurs différentes de la constante de vitesse pour les deux états B et C. Ils les estiment respectivement à (8,8 ± 1,5).10-10 cm3s-1 et (3,3 ± 0,3).10-10 cm3s-1. Il s’agit d’une mesure directe du processus de destruction par collision binaire avec Cl2 qui inclut tous les phénomènes et pas seulement le quenching. En effet, comme les états B et C sont proches énergétiquement, on peut penser qu’il y a un processus de couplage collisionnel des deux états. Un résultat analogue pour le xénon semble renforcer cette hypothèse sur laquelle nous reviendrons au paragraphe suivant.
Des atomes de chlore libres existent aussi dans les conditions qui règnent dans les lasers. La réaction du quenching suivante est prévue : XeCl* + Cl → Xe + 2Cl
La constante de vitesse a été estimée par deux auteurs : 1,4.10-9 cm3s-1[124] et 8.10-10 cm3s-1[140].
La présence d’impuretés Im telles que les chlorocarbones (qui sont la conséquence de phénomènes de corrosion[186]), NO, CO2, O2, CO, N2O, H2O pourraient influer sur la cinétique de disparition de XeCl* puisque les collisions binaires Im–XeCl* possèdent des constantes de vitesse de l’ordre de 3.10-10 cm3s-1[172] donc comparables à celle de la réaction XeCl* + RCl. Cependant, compte tenu des teneurs usuelles en impuretés, les fréquences des réactions sont négligeables. Une solution technique a été proposée pour les éliminer. Elle consiste à introduire 1 torr de H2[186].
Dans les mélanges binaires Xe/HCl
Compte tenu du faible écart énergétique (environ 100 cm−1) entre ces deux états (tableau 2), on peut penser qu’un couplage se produit. Nous avons vu au paragraphe précédent que le chlore Cl2 pouvait jouer un rôle dans les processus de couplage. Toutefois, ce résultat n’a pas été chiffré exactement ni confirmé par la suite. En effet, aucun phénomène de couplage collisionnel induit par le donneur de chlore n’a été décelé dans les études les plus récentes.
Le rôle des électrons n’est pas bien connu dans ce processus de couplage. Pour Finn et al.[169] il est négligeable. Ce n’est pas le cas pour Johnson et al.[140] qui donnent une constante de vitesse élevée. Celle-ci est selon eux identique pour le transfert de B vers C et pour celui de C vers B. Une hypothèse bien peu vraisemblable qui a pour conséquence que la différence d’énergie entre B et C est nulle. Ce qui n’est pas le cas (tableau 2). Ils estiment le taux de réaction à 2.10-8 cm3s-1.
Le couplage se manifeste grâce à des collisions binaires avec un atome de xénon :
XeCl(B ; v’ = 0) + Xe → XeCl(C ; v’ = 0,1) + Xe (15)
avec la constante de vitesse kBC
XeCl(C ; v’ = 0, 1) + Xe → XeCl(B ; v’ = 0) + Xe (16)
avec la constante de vitesse kCB
Les mesures des constantes de vitesse ne sont pas très concordantes comme on peut le constater sur le tableau 28.
Tableau 28 : Constantes de vitesse en cm3s-1 des processus de couplage collisionnel des états B et C.
Dans les expériences d’Inoue et al.[66], les niveaux vibrationnels v’ = 0,1 sont excités directement. Ce n’est pas le cas dans les autres expériences[22] - [76]. La dernière valeur[126] n’est qu’une estimation théorique basée sur des analogies avec d’autres réactions. Si l’on s’intéresse à l’écart énergétique ΔE = EB - EC qui est déduit de kCB et kBC, on peut avoir plus de renseignements sur la valeur des mesures. Si l’on suppose que le états EB et EC sont thermalisés :
kBC/kCB = exp(ΔE/kT)
car les poids statistiques des deux états sont les mêmes[54].
ΔE ainsi déduit de Inoue et al.[66] vaut 85 cm−1 et 119 cm−1 pour Rives et al.[22], alors que l’on obtient –22 cm−1 avec les mesures de Le Calvé et al.[76] (voir tableau 2). Seules les deux premières sont donc une valeur de ΔE compatible avec l’ordre de grandeur couramment admis (100 cm−1). Une nette différence demeure entre ces deux travaux : un ordre de grandeur sépare les valeurs de kBC et kCB dans les deux expériences[66] - [22]. Grieneisen et al.[72] donnent seulement les taux de destruction globaux des états B et C, c’est-à-dire quenching + couplage. Ils trouvent pour la destruction de l’état C (15,5 ± 0,9).10-12 cm3s-1 et pour l’état B (10,3 ± 0,9).10-12 cm3s-1, c’est-à-dire des valeurs intermédiaires entre celles de Inoue et al.[66] et Rives et al.[22]. Rappelons-nous que le quenching par le xénon n’a qu’une faible influence (tableau 20). Soulignons enfin que Inoue et al.[66] ne tient pas compte de la réaction (9). Si l’on fait de même dans les résultats de Rives et al.[22], les valeurs de kBC et kCB se rapprochent de celles de Inoue et al.[66]. Comme pour kx et kH, la prise en compte du processus (9) modifie les valeurs des taux de réaction. Sur ce point Rives et al.[22] sont plus précis qu'Inoue et al.[66].
L’avantage très net de Inoue et al.[66] est la résolution vibrationnelle dans leurs mesures. En effet, kBC et kCB varient avec le niveau vibrationnel v. Pour les niveaux v = 70 à 130, on a observé[168] des constantes de vitesse qui sont comprises entre 15 et 20.10-11 cm3s-1. kBC et kCB semblent donc croître avec v.
Comme XeCl(B, C) se forme la plupart du temps avec une forte excitation vibrationnelle, il est donc important de savoir deux choses : la première, l’estimation exacte de la variation de kBC et kCB avec v ; la seconde, connaître la cinétique de relaxation vibrationnelle et son importance relative vis-à-vis des processus de couplage. Ce sera l’objet des deux prochains paragraphes.
Rôle du gaz tampon
Le couplage collisionnel se produit par collision binaire avec un atome de gaz rare Rg :
XeCl(B) + Rg → XeCl(C) + Rg (17)
avec la constante de vitesse kBCRg
XeCl(C) + Rg → XeCl(B) + Rg (18)
avec la constante de vitesse kCBRg
Les résultats que nous présentons sont ceux de Dreiling et Setser[168]. Ils ne donnent pas les valeurs exactes de kBCRg et kCBRg mais un ordre de grandeur pour un intervalle donné de niveaux vibrationnels. Leurs résultats sont présentés dans le tableau 29. Ils montrent que les valeurs des constantes de vitesse augmentent régulièrement lorsque le niveau vibrationnel v de XeCl* est plus élevé et que le gaz rare Rg est plus lourd.
v He Ne Ar Kr 0–30 (0,5 à 1,8) × 10-11 (0,7 à 2,6) × 10-11 (3,0 à 11) × 10-11 (3,0 à 11) × 10-11 30 – 70 (1,8 à 2,5) × 10-11 (2,6 à 3,5) × 10-11 (11 à 15) × 10-11 (11,0 à 16) × 10-11 70 - 130 2,5 × 10-11 3,5 × 10-11 15 × 10-11 16 × 10-11
Tableau 29 : Constantes de vitesse de couplage en cm3s-1 par collisions binaires avec un atome gaz rare[168].
On dispose de quelques éléments de comparaison.
Dans l’hélium, des expériences ont été menées à basse et haute pression[71]. À haute pression, les constantes de transfert sont de l’ordre de (1,5 ± 0,7).10-12 cm3s-1 et à basse pression 3,0.10-11 cm3s-1. Ces résultats sont en accord avec les précédents. En effet, une forte pression induit une relaxation vibrationnelle donc les valeurs de v concernées par le transfert sont faibles et vice-versa pour les faibles pressions. La seule détermination directe disponible de kBCHe donne une valeur inférieure à 3.10-13 cm3s-1[74].
Pour le néon, les valeurs des taux de transfert mesurées à basse et haute pression sont respectivement 3,0.10-11 cm3s-1 et (0,8 ± 0,4).10-12 cm3s-1[71]. Elles sont inférieures à celles du tableau 29. La mesure directe de la constante de vitesse kBCNe donne une valeur inférieure à 3.10-13 cm3s-1[74]. Enfin, selon un autre auteur[161], l’ordre de grandeur de deux constantes de vitesse de couplage serait de 4,8.10-12 cm3s-1 pour v = 4.
Pour l’argon, il y a plus de résultats. À basse pression, l’ordre de grandeur ne serait que de 6,0.10-11 cm3s-1[71]. D’autres auteurs[70] annoncent pour les taux de transfert (1,2 ± 0,4).10-4 cm3s-1 pour un intervalle de pression allant de 10 à 1 000 torr. Des mesures directes de kBCAr et kCBAr sont disponibles sans toutefois préciser les niveaux vibrationnels en cause[55] :
kBCAr = 36.10-4 cm3s-1 et kCBAr = 21.10-11 cm3s-1
Yu et al.[74] notent quant à eux une variation de kBCAr avec la température :
kBCAr = (4 ± 2).10-12 cm3s-1 à 300K et kBCAr = (2 ± 1).10-12 cm3s-1 à 230K
Pour le krypton, on ne connaît qu’une estimation :
kBCKr = (4).10-12 cm3s-1[74]
Il est clair que les processus de couplage collisionnels induits par les gaz rares sont très mal connus. Ils varient de plusieurs ordres de grandeurs entre les différents auteurs. L’incertitude sur les constantes de vitesse est donc aussi importante que pour le xénon. L’excitation vibrationnelle semble jouer un rôle qui n’est pas encore bien élucidé. Les résultats disponibles sont fragmentaires et il manque des mesures directes de kBCRg et kCBRg. À partir des premières estimations, ces phénomènes semblent importants dans la cinétique des mélanges gazeux et il convient d’encourager à mieux les connaître.
Relaxation vibrationnelle
Comme nous l’avons vu XeCl* se forme le plus souvent avec une forte excitation vibrationnelle qui peut atteindre v = 100[187]. Cela induit des phénomènes de relaxation vibrationnelle qui se font essentiellement par collision binaire avec un atome de gaz rare[188].
Nous ne disposons que d’une seule mesure pour le xénon ; elle est relative au niveau v = 2.
XeCl(B ; v = 2) + Xe → XeCl(B ; v’ = 0,1) + Xe
avec la constante de vitesse kv
avec kv = (2 ± 1).10-10 cm3s-1[66].
L’essentiel des résultats connus sont relatifs aux gaz tampons. C’est encore les travaux de Dreiling et Sester[168] qui sont les plus complets. La relaxation vibrationnelle s’écrit :
XeCl*(v) + Rg → XeCl*(v’) + Rg (19)
Les ordres de grandeur de kvRg sont rassemblés dans le tableau 30. On y constate que kvRg augmente avec le niveau vibrationnel de XeCl* et si le gaz rare Rg est de plus en plus lourd. Les valeurs de kvRg sont censées être les mêmes pour les états B et C.
v He Ne Ar Kr 0–30 (0,15 à 1,1) × 10-11 (0,5 à 2,9) × 10-11 (1,0 à 6,0) × 10-11 (0,6 à 2,7) × 10-11 30 – 70 (1,1 à 2,5) × 10-11 (2,9 à 6,2) × 10-11 (6,0 à 12) × 10-11 (2,7 à 5,5) × 10-11 70 - 130 (2,5 à 4,4) × 10-11 (6,2 à 9,5) × 10-11 (20 à 34) × 10-11 (5,5 à 7,3) × 10-11
Tableau 30 : Constantes de vitesse de relaxation vibrationnelle en cm3s-1 induites par collisions binaires avec un atome de gaz tampon Rg[168].
Pour l’hélium et le krypton, nous ne disposons d’aucune base de comparaison.
Pour le néon, seule la réaction concernant les deux premiers niveaux vibrationnels de B a été chiffrée :
XeCl(B ; v = 1) + Ne → XeCl(B ; v = 0) + Ne
avec la constante de vitesse kvNe=(0,3 à 0,5).10-11 cm3s-1[189].
Pour l’argon, les valeurs de kvAr ont été déterminées pour v = 33, 60 et 75[95]. Elles valent respectivement (17 ± 5).10-11 ; (31 ± 9).10-11 et (43 ± 10).10-11 cm-11. D’autres auteurs chiffrent kvAr entre (10 et 15).10-11[160]. Notons qu’il y a simplement accord sur l’ordre de grandeur.
Conclusion à propos des voies de disparition de l’exciplexe
Il apparaît clairement que la cinétique relative au couplage collisionnel des états B et C et la relaxation vibrationnelle sont encore mal connues. Le peu de résultats disponibles ne sont pas souvent en accord. Par contre, on a une bonne idée générale de la situation. On sait que pour les niveaux vibrationnels élevés, le couplage l’emporte sur la relaxation vibrationnelle et pour les niveaux les plus bas, c’est le contraire[63], ceci quel que soit le gaz rare impliqué dans la réaction.
Il peut être intéressant de comparer l’importance relative des différents processus de destruction de l’exciplexe XeCl(B) qui est l’état de départ de la transition laser. Pour cela, nous choisissons un mélange type utilisé dans les lasers. C’est le néon qui est le plus souvent choisi comme gaz tampon. L’argon est de plus en plus rejeté à cause de la forte absorption de l’ion Ar2+ à 308 nm[140]. Le mélange sera donc ternaire (Ne/Xe/HCl). La pression totale sera fixée à 3 atm, les pressions partielles respectives seront 2268,6 torr, 10 torr et 1,4 torr. Les constantes de vitesse sont les valeurs moyennes des estimations les plus fiables.
Les résultats sont rassemblés sur le tableau 31. Pour ce qui est de la réaction (19) nous n’avons pris en compte que les plus bas niveaux vibrationnels. Ainsi 0,40 ns-1 représente une fréquence limite inférieure de disparition. Il s’agit d’ailleurs du processus qui induit la plus forte destruction. Ceci indique que XeCl(B) formé avec une forte excitation vibrationnelle, est rapidement relaxé par collision binaire avec le néon mais aussi vraisemblablement par le xénon. On peut donc penser que les autres processus ne sont réellement perceptibles qu’une fois XeCl(B) sur le niveau v = 0. C’est la raison pour laquelle, pour la réaction (17), nous avons choisi la valeur de kBCNe relative à v faible. Une fois la relaxation terminée les autres processus prennent le relais. La dépopulation par émission spontanée est très importante ainsi que les réactions (11) et (17). Or pour ces deux processus nous avons vu qu’il y avait une forte incertitude sur les mesures en même temps qu’un faible nombre de déterminations. Le rôle du couplage par le xénon n’est pas mieux connu mais il a une influence moindre de même que la destruction par collision binaire avec HCl. Les autres processus mieux connus, eux, sont paradoxalement négligeables. On y trouve en particulier toutes les réactions à trois corps.
Processus Voie radiative 6 7 8 9 11 12 13 15 17 19 Fréquence 0,099 0,036 0,001 0,0001 0,0008 0,24 0,0006 0,0074 0,027 0,064 0,40 Pourcentage 11 % 4 % < 1 % < 1 % < 1 % 27 % < 1 % 1 % 3 % 7 % 46 % Pourcentage après relaxation vibrationnelle 21 % 8 % < 1 % < 1 % < 1 % 50 % < 1 % 2 % 6 % 13 %
Tableau 31 : Fréquences de destruction des états B en ns-1.
Il semble donc nécessaire de préciser la cinétique car les processus les plus mal connus sont ceux qui à première vue jouent un rôle important : relaxation vibrationnelle, couplage collisionnel, tous partenaires confondus.
L’exciplexe Xe2Cl
D’une manière générale les molécules Rg2X sont moins stables que les RgX[9]. Dans notre cas, Xe2Cl a un double intérêt. Il peut perturber les performances du laser XeCl car il absorbe beaucoup à 308 nm et par contre il permet aussi le développement d’un autre type de laser basé sur une émission de Xe2Cl.
La molécule Xe2Cl
D’après les premiers travaux effectués sur cette molécule[39] - [190], on note les points suivants :
- la configuration la plus stable de la molécule dans un état excité est une géométrie triangulaire C2v ;
- les états excités Xe2Cl* sont des complexes résultant de l’association d’un ion moléculaire Xe2+ et d’un ion atomique Cl− ;
- l’émission observée de la molécule est large ; les transitions correspondantes aboutissent sur une partie très répulsive de l’état fondamental.
Les courbes de potentiel calculées par Huestis et al.[191] selon la méthode DIM (Diatomics In Molecules) sont présentées sur la figure 15.
Les trois états les plus bas sont covalents et répulsifs. Ils sont corrélés à XeCl(X ou A) et à un atome de xénon à l’état fondamental. La valeur expérimentale de l’énergie de l’état 12Γ est de 0,273 eV[39]. Elle est compatible avec ces courbes de potentiel. Les trois états suivants sont ioniques. L’état lié 42Γ est corrélé à XeCl(B) + Xe ; le suivant 52Γ, répulsif, est corrélé à XeCl(C) + Xe.
Plus récemment, Last et George[49] ont déterminé les courbes de potentiel à l’aide d’une autre méthode, la méthode DIIS (Diatomics In Ionic Systems) sans prise en compte du couplage spin-orbite. Ils trouvent comme Huestis et al.[191] que l’état 42Γ est l’état ionique le plus bas. Au fond du puits, cet état possède la configuration d’un triangle isocèle tel que la distance entre les positions d’équilibre de Xe et Cl vaut 3,23 Å. Pour Adams et Chabalowski[48] la distance Xe–Cl vaut 3,39 Å.
Dans un premier temps, les courbes de potentiel des différents états ont été tracées en maintenant constante et égale à 3,25 Å la distance Xe–Xe (figure 16). Last et George obtiennent neuf états (trois covalents et six ioniques). On remarque que les courbes potentielles des états antisymétriques 42Γπ et 62Γπ coïncident presque avec les potentiels des états symétriques 52Γ et 62Γ. Les états 32Γ et 72Γ mis en évidence de par Huestin et al.[191] sont absents du fait de la non prise en compte du couplage spin-orbite. À l’inverse, trois états (22Γπ, 42Γπ et 62Γπ) qui ont la symétrie π ne figurent pas dans les diagrammes donnés par Huestis et al.[191].
Le même travail a été repris dans un deuxième temps en maintenant constante la distance Xe-Cl à 3,23 Å (figure 17).
On peut tirer d’autres résultats de l’article de Last et George[49] :
1) dans l’état 42Γπ la molécule à la configuration d’un triangle isocèle tel que les distances Xe-Cl et Xe–Xe valent respectivement 3,13 et 4,23 Å. L’état est à 0,8 eV au-dessus de l’état 42Γ.
2) l’état fondamental 12Γ est un complexe de Van der Walls. Il possède une énergie de dissociation de 0,075 eV et une configuration triangulaire dissymétrique. Les distances Xe–Cl sont de 3,23 et 4,06 Å et l’angle Xe–Cl–Xe vaut 74,4°.
3) le premier état excité 22Γ est aussi un complexe de Van der Walls. Il a une géométrie symétrique avec une distance Xe–Cl de 3,99 Å et un angle Xe–Cl–Xe de 68,4°. Son énergie de dissociation est évaluée à 0,055 eV.
Toujours selon Last et George[49], il existe un autre mode de description de la molécule dont la configuration à l’état stable est linéaire et symétrique (Xe–Cl–Xe). Pour l’état fondamental, la distance Xe–Cl serait de 3,24 Å et l’énergie de dissociation de 0,076 eV. Il pourrait aussi exister un état excité correspondant à une distance Xe-Cl de 3,06 Å. Cet état non représenté sur les figures 16 et 17, posséderait une énergie supérieure à 0,72 eV à celle de l’état 42Γ. La liaison serait là aussi de type ionique.
Seule une expérience menée à l’état solide[77] peut être comparée à ces travaux théoriques. L’unique état étudié est le 42Γ. La structure de triangle isocèle de cet état y est confirmée. Trois grandeurs peuvent être confrontées aux prédictions théoriques. La distance Xe–Xe est mesurée à 3,17 Å et celle de Xe–Cl à 3 Å. L’accord est meilleur pour l’énergie du fond du puits évaluée à 3,15 eV. Par ailleurs, les auteurs ont déterminé les fréquences fondamentales vibrationnelles pour chacune des liaisons. Pour Xe–Xe, ωx = 123 cm−1 et pour Xe–Cl, ωc = 180 cm−1.
Voies de formation
Trois voies principales de formation de Xe2Cl* sont énergétiquement possibles par collision et deux autres par photoassociation :
Xe2*(A1Σ) + Cl2 → Xe2Cl* + Cl (20)
Xe* + Xe + Rg → Xe2Cl* + Rg (21)
Xe2+ + Cl− + Rg → Xe2Cl* + Rg (22)
XeCl*(X) + Xe + hν → Xe2Cl* (23)
Xe + Cl + Xe + hν → Xe2Cl* (24)
où Rg désigne un gaz rare qui peut être du xénon ou un gaz tampon.
Les auteurs sont en désaccord sur l’importance relative de ces processus de formation. Elle dépend des conditions expérimentales. Examinons les caractéristiques de chacune d’entre elles.
Par harponnage
La réaction (20) est une réaction « harpon » très énergétique. Elle fait intervenir des états excités Xe2*. Pour Bruce et al.[117], c’est la voie de formation dominante. Ce point de vue n’est pas partagé par d’autres auteurs qui considèrent la réaction comme faible[191] voire négligeable[192]. Aucune constante de vitesse n’a encore été mesurée.
Voie photoassociative
Les réactions (23) et (24) n’ont été mises en évidence que très récemment[111]. Aucune information nouvelle n’a été publiée.
Voie ionique
Selon un calcul théorique[152], le taux de recombinaison α’ des ions Xe2+ et Cl− lorsque Rg = Xe (réaction (22)) a été estimé une première fois à 1.10–7cm3.s-1. Les mêmes auteurs viennent de réviser à la baisse cette valeur : α’ = 5.10–8cm3.s-1[193]. Ce résultat est conforté expérimentalement[191] - [194]. Notons que selon les calculs, cette réaction peut devenir importante à très forte pression. Xe2Cl* devient alors le principal produit de la réaction, ceci au détriment de XeCl* (réaction (4)).
Réactions ternaires
Beaucoup d’auteurs considèrent que la formation de Xe2Cl* est assurée principalement par la voie (21). D’après une étude récente[67], la réaction peut être interprétée comme le résultat de deux réactions successives, la seconde correspondant à une relaxation vibrationnelle par collision avec Rg :
XeCl(B,C) + Xe ↔ Xe2Cl*(v)
Xe2Cl*(v) + Rg → Xe2Cl* + Rg
D’après les auteurs, les niveaux vibrationnels de départ Xe2Cl*(v) sont au-dessus de la limite de dissociation de l’état en XeCl* + Xe.
Par contre, pour Yu et al.[74], la création de Xe2Cl* passe par celle d’un complexe triatomique RgXeCl*, soit :
XeCl* + Rg → RgXeCl* où Rg≠Xe
RgXeCl* + Xe → Xe2Cl* Rg
Ces auteurs n’ont observé ces réactions que dans le cas de l’argon et du krypton.
La seconde réaction est une réaction de déplacement. Une autre réaction lui est compétitive lorsque le xénon est remplacé par le krypton. Ce processus de quenching aurait une constante de vitesse légèrement supérieure à 1.10-13cm3.s-1[74] - [182].
La durée de vie du complexe RgXeCl* est mal connue. Elle est estimée à 200 ns pour KrXeCl[74] - [182] et 40 ns pour NeXeCl[96]. Cet intervalle de temps est suffisamment long pour que la deuxième collision ait donc une chance raisonnable de se produire.
Quoi qu’il en soit, les constantes de vitesse de (21) ont été mesurées et elles sont rassemblées dans le tableau 32. Si Rg≠Xe, seules deux mesures directes ont été effectuées[44] - [67]. La dernière[195]n’est qu’une évaluation.
Tableau 32 : Constantes de vitesse en cm6s-1 de la réaction (21).
Dans le cas du xénon, notons que l’ensemble des constantes kDX du tableau 20 peuvent être considérées comme la cinquième colonne du tableau 32 puisque kDX peut se confondre avec la réaction (21)[67].
Spectres d’émission
Les études théoriques[163] - [190] montrent que les transitions permises sont les suivantes (figure 15) :
42Γ → 12Γ (A)
42Γ → 22Γ (B)
42Γ → 32Γ (C)
L’état de départ est toujours le même et les longueurs d’onde λTh correspondantes sont indiquées dans le tableau 33. On peut les comparer aux valeurs expérimentales λObs.
Tableau 33 : Caractéristiques des émissions de Xe2Cl*.
Expérimentalement, Fajardo et Apkarian[77] ont pu observer deux transitions dans le domaine spectral considéré, qui d’après eux sont les transitions (A) et (B), même s’il y a un décalage important en longueur d’onde. En fait, dans la plupart des cas, on observe un continuum très large (environ 80 nm) qui recouvre les trois émissions. Le positionnement du maximum varie beaucoup selon les auteurs ; les valeurs oscillent entre 450 et 500 nm. Un exemple de spectre est donné sur la figure 11. La limite de l’émission du côté des courtes longueurs d’onde a été évaluée par le calcul à 443 nm[106].
D’après Last et George[49], la molécule linéaire Xe–Cl–Xe devrait produire une émission vers l’état fondamental à 321 nm et le moment de transition serait élevé : 3,9 D. Pour autant, il n’y a à ce jour aucune confirmation expérimentale de cette prévision.
À l’état solide, l’émission de Xe2Cl* est très décalée vers le rouge. Elle est centrée aux environs de 570 nm[196] - [197]. On observe un résultat analogue à l’état liquide[198]. Selon ces auteurs, ce phénomène serait dû à la déformation des courbes de potentiel découlant des interactions entre molécules qui sont plus proches les unes des autres qu’à l’état gazeux. Une étude théorique[199] explique pour sa part ce phénomène par la polarisation de la matrice de xénon par Xe2+Cl− ainsi qu’une contribution importante des forces de Van der Walls.
L’émission du trimère Xe2Cl* n’est observée qu’à forte pression de gaz rare (xénon ou gaz tampon) et la fluorescence augmente avec la pression de xénon[39]. Ce résultat peut s’expliquer si l’on considère que la voie de formation de Xe2Cl* correspond à la réaction (21). Compte tenu des valeurs des constantes de vitesse des réactions de type (21), la fréquence de la réaction ne devient significative que lorsque la pression de gaz rare est supérieure à environ 200 torr. Quant à la réaction (22), elle n’intervient que lorsque la pression est de plusieurs atmosphères[193].
Durée de vie de Xe2Cl (42Γ)
Nous venons de voir que le seul état de Xe2Cl à être à l’origine d’une émission lumineuse est 42Γ). De nombreuses déterminations de sa durée de vie ont été effectuées. Les résultats obtenus à l’état gazeux sont rassemblés dans le tableau 34. Les résultats sont très variables et les incertitudes importantes. L’intervalle de confiance obtenu au seuil de 5 % est compris entre 240 et 253 ns. Seules quatre valeurs en font partie[67] - [85] - [172] - [194]. Remarquons que compte tenu de sa forte incertitude absolue, une autre mesure[116] a un intervalle commun avec l’intervalle de confiance.
Durées de vie (ns) Références 300 ± 50 Berman[116] 185 ± 10 Grieneisen et al.[72] 260 Lorents[75] 135+70-60 Marowsky et al.[44] 210 ± 25 Marowsky et al.[200] 250 ± 25 Mc Cown et al.[194] 245 ± 10 Mc Cown et Eden[85] 328 ± 20 Okada et Apkarian[201] 250 Quiñones et al.[67] 330 Stevens et Krauss[163] 210 ± 20 Tang et al.[170] 242 ± 10 Yu et al.[172]
Tableau 34 : Durées de vie de Xe2Cl(42Γ) obtenues expérimentalement à l’état gazeux sauf pour la référence Stevens et Krauss[163] qui est une détermination théorique.
Les mesures réalisées à l’état solide fournissent des valeurs encore plus dispersées comme le montre le tableau 35.
Tableau 35 : Durées de vie de Xe2Cl(42Γ) observées à l’état solide.
Rôle des donneurs de chlore (RCl)
Outre la désexcitation radiative, l’état Xe2Cl(42Γ) est détruit par collision double avec RCl. Pratiquement, tous les auteurs s’accordent pour dire qu’il s’agit de la voie dominante de disparition de Xe2Cl* par processus collisionnel, et ceci quel que soit le donneur de chlore. On comprend dans ces conditions que les émissions de Xe2Cl* ne soient observées que pour des concentrations de RCl très faibles[21] - [117] - [172]. Les valeurs des constantes de vitesse des réactions (24) sont données dans le tableau 36.
Xe2Cl* + RCl → produits autres que Xe2Cl (24)
Références Cl2 HCl CCl4 Marowsky et al.[200] (2,2 ± 0,2) × 10-10 (4,3 ± 0,4) × 10-10 (5,4 ± 0,5) × 10-10 Lorents[75] (6,1 ± 0,2) × 10-10 Baginskii et al.[157] 2,6 × 10-10 Tang et Lorents[202] 8 × 10-10 Kannari et al.[126] 6,1 × 10-10 Turner et Smith[135] 6 × 10-10 Berman[116] (3,9 ± 0,4) × 10-10 Tang et al.[170] (4,5 ± 0,4) × 10-10 Grieneisen et al.[72] (2,6 ± 0,3) × 10-10 Mc Cown et Eden[85] (4 ± 1) × 10-10 Okada et Apkarian[201] (7 ± 1) × 10-10 Yu et al.[172] (4,0 ± 1) × 10-10-10 Zuev et al.[203] 1,8 × 10-10 Marowsky et al.[44] (6 ± 1) × 10-10
Tableau 36 : Constantes de vitesse en cm3s-1 des réactions (24) pour divers donneurs de chlore RCl.
Il n’y a que deux déterminations pour CCl4 mais elles coïncident. Dans le cas de HCl deux valeurs sont statistiquement éloignées des autres[157] - [202] et il est difficile d’avancer une explication à cette situation. L’intervalle de confiance au seuil de 5 % est de 4 à 7.10-10 cm3s-1.
Dans le cas du chlore Cl2, seule la moitié des mesures sont statistiquement proches[85] - [116] - [170] - [172]. Là encore, il est difficile d’expliquer les écarts observés. L’intervalle de confiance au seuil de 5 % varie de 3,7 à 4,5.10-10 cm3s-1.
On peut donc conclure que les trois donneurs de chlore ont une influence analogue sur la destruction collisionnelle de Xe2Cl*.
Il existe aussi une estimation de la constante de vitesse de la réaction :
Xe2Cl* + Cl → 2 Xe + 2 Cl
Elle vaut : 1.10-9 cm3s-1[204].
Rôle des gaz rares
Il s’agit uniquement de réactions binaires :
Xe2Cl* + Rg → produits autres que Xe2Cl (25)
La disparition de Xe2Cl* par collision sur un atome de xénon a été observée par Grieneisen et al.[72] ; la constante de réaction vaut 6.10-15 cm3s-1. Pour les autres auteurs, cependant, cette réaction ne peut être mise en évidence[44] - [75] - [170] - [201] - [203]. Pour l’instant, la limite supérieure de la constante de vitesse de la réaction (25) est 1.10-17 cm3s-1[201]. D’autres auteurs, placent cette limite plus haut 4 à 7.10-14 cm3s-1[170] - [203] ou 5.10-13 cm3s-1[44]. Notons que la valeur retenue par Kannari et al.[126] qui est 8.10-12 cm3s-1 n’a aucun fondement.
Dans le cas des mélanges ternaires, le rôle du gaz tampon est mal connu.
Dans le cas de l’argon, nous avons deux mesures : (3 ± 1).10-14 cm3s-1[44] et (1,5 ± 0,4).10-14 cm3s-1[200].
Pour l’hélium, deux valeurs ont été avancées : 5.10-13 cm3s-1[157] et 3.10-14 cm3s-1[124].
Rôle des électrons et des impuretés
La réaction (26) a fait l’objet de plusieurs estimations qui sont très peu en accord. Elles sont rassemblées dans le tableau 37.
Xe2Cl* + e− → 2 Xe + Cl + e− (26)
Tableau 37 : Constantes de vitesse en cm3s-1 de la réaction (26).
D’après Yu et al.[172], les impuretés ont une influence moindre dans la cinétique de disparition de Xe2Cl* que dans celle de XeCl*. Les constantes de vitesse de disparition à deux corps Im–Xe2Cl* sont en effet d’un ordre de grandeur plus faible que les constantes relatives aux collisions binaires Im–XeCl*. Toutefois, pour CO2 et NO, les constantes de vitesse sont du même ordre de grandeur (quelque 10−10 cm3 s−1) que celles du type RCl–Xe2Cl*. Il faut bien entendu tenir compte des teneurs en impuretés qui sont le plus souvent très faibles. Les fréquences de réaction sont alors négligeables.
Conclusion
Malgré tous les travaux effectués sur les chlorures de xénon, beaucoup de zones d’ombres et d’incertitudes demeurent quant à la structure des molécules et aux processus collisionnels.
Si les longueurs d’onde des émissions qu’ils produisent sont connues avec précision, la cinétique de disparition des états excités est encore souvent imprécise. C’est le cas des processus de relaxation vibrationnelle de XeCl(B, C), de l’influence des gaz tampons notamment pour ce qui est des constantes de vitesse de couplage collisionnel entre les états B et C de XeCl, et aussi des collisions tertiaires. Parce qu’elle est bien moins complexe que de XeCl*, la cinétique de disparition de Xe2Cl* est mieux comprise.
Ces réactions mal connues jouent pourtant un rôle fondamental dans la cinétique du laser XeCl. La modélisation du milieu gazeux devrait être influencée par leur remise en question. Malgré tout quelques mesures directes expérimentales récentes sont disponibles et il convient de les préférer à des estimations hasardeuses et approximatives. La complexité des voies de formation de XeCl a aussi été mise en lumière. Même si les processus de harponnage jouent un rôle secondaire, ils n’en demeurent pas moins non négligeables. Des nouvelles voies de formation ont été récemment mises en évidence et leur importance apparaît qualitativement comme de première grandeur. Autant de pistes nouvelles qu’il convient d’analyser en détail.
Notes et références
- Masse molaire calculée d’après « Atomic weights of the elements 2007 », sur www.chem.qmul.ac.uk.
- (en) Hasso Meinert, « Über die Bildung von Xenondichlorid », Zeitschrift für Chemie, vol. 6, no 2, , p. 71–71 (DOI 10.1002/zfch.19660060210, lire en ligne, consulté le ).
- « Author index », Endeavour, vol. 23, no 90, , ii (ISSN 0160-9327, DOI 10.1016/0160-9327(64)90050-x, lire en ligne, consulté le ).
- A.V. Eletskii, Sov. Phys. Usp. 21, 502 (1978).
- M.J. Shaw, Prog. Quant. Electr. 6, 3 (1979).
- Excimer lasers (Springer–Verlag, Berlin, 1979) edited by Ch. K. Rhodes.
- M.H.R. Hutchinson, Appl. Phys. 21, 95 (1980).
- I.S. Lakoba and S.I. Yakovlenko, Sov. J. Quantum Electron. 10, 389 (1980).
- B.M. Smirnov, Sov Phys. Usp. 26, 31 (1983).
- APS Study : Science and Technology of Directed Energy Weapons, Rev. Mod. Phys. 59, S9 (1987).
- F.K. Tittel, G. Marowsky, W.L. Wilson Jr., and M.C. Smayling, IEEE J. Quantum Electron. QE-17, 2268 (1981).
- A. Garscadden, M.J. Kushner, and J.G. Eden, IEEE Trans. Plasma Sci. 19, 1013 (1991).
- M.R. Flannery, Int. J. Quantum Chem. 13, 501 (1979).
- B.L. Fontaine, B.M. Forestier, M. Sentis, P. Delaporte, and L. Arif, J. Phys. Colloq. 48, C7-331 (1987).
- I.A. Mc Intyre and C. K. Rhodes, J. Appl. Phys. 69, R1 (1991).
- A. Belasri, Thèse de doctorat de l’Université Paul Sabatier, no 1607, Toulouse (1993).
- V. Baudinaud and M. Autric, Ann. Phys. Colloq. 17, 1 (1992).
- S. Avrillier, E. Tinet, and D. Ettori, Ann. Phys. Colloq. 17, 13 (1992).
- J. Bretagne and E. Estocq, Ann. Phys. Colloq. 17, 29 (1992).
- M. Sentis, Entropie (Paris) 20, 3 (1984).
- H. Asselman, P. Rives, J. Galy, H. Brunet, and J.L. Teyssier, J. Phys. B : At. Mol. Opt. Phys. 26, 2311 (1993). .
- P. Rives, J.L. Teyssier, J. Galy, A. Briot, H. Brunet, and H. Asselman, J. Chem. Phys. 102, 1217 (1995). .
- H. Asselman, A. Sekaki, J. Galy, P. Rives, H. Brunet, A. Birot, and J.L. Teyssier, Appl. Radiat. Isot. 46, 475 (1995). .
- P. Rives, Thèse de doctorat de l’Université Paul Sabatier, no 1505, Toulouse (1993).
- A.P. Golovitskii, Sov. Tech. Phys. Lett. 18, 269 (1992).
- G.B. Rulev and V.B. Saenko, Tech. Phys. Lett. 19, 687 (1993).
- A. Von Antropoff, Z. Angew. Chem. 37, 217 (1924).
- G. Oddo, Gazz. Chem. Ital. 63, 380 (1933).
- D.M. Yost and A. L. Kaye, J. Am. Chem. Soc. 55, 3890 (1933).
- H. Meinert, Z. Chem. 6, 71 (1965).
- D.M. Proserpio, R. Hoffmann, and K.C. Janda, J. Am. Chem. Soc. 113, 7184 (1991).
- J.M. Riveros, P.W. Tiedemann, and A.C. Breda, Chem. Phys. Lett. 20, 345 (1973).
- J.E. Velazco and D.W. Setser, J. Chem. Phys. 62, 1990 (1975).
- J.J. Ewing and C.A. Brau, Appl. Phys. Lett. 27, 350 (1975).
- W.J. Stevens and M. Krauss, J. Chem. Phys. 77, 1368 (1982).
- R.W. Waynant and J.G. Eden, Appl. Phys. Lett. 36, 262 (1980).
- R.C. Sze and P.B. Scott, Appl. Phys. Lett. 33, 419 (1978).
- W.L. Nighan and R.T. Brown, Appl. Phys. Lett. 36, 498 (1980).
- D.C. Lorents, D.L. Huestis, M.V. Mc Cusker, H.H. Nakano, and R.M. Hill, J. Chem. Phys. 68, 4657 (1978).
- K.Y. Tang, D.C. Lorents, and D.L. Huestis, Appl. Phys. Lett. 36, 347 (1980).
- F.K. Tittel, W.L. Wilson, R.E. Stickel, G. Marowsky, and W.E. Ernst, Appl. Phys. Lett. 36, 405 (1980).
- M.E. Fajardo and V.A. Apkarian, Chem. Phys. Lett. 134, 51 (1987).
- L. Wiedeman, M.E. Fajardo, and V.A. Apkarian, Chem. Phys. Lett. 134, 55 (1987).
- G. Marowsky, E.P. Glass, M. Smayling, F.K. Tittel, and W.L. Wilson, J. Chem. Phys. 75, 1153 (1981).
- K.V. Chance, K.H. Bowen, J.S. Win, and W. Klemperer, J. Chem. Phys. 70, 5157 (1979).
- E.W. Boom and J. Van der Elsken, J. Chem. Phys. 73, 15 (1980).
- I. Last and T.F. George, J. Chem. Phys. 89, 3071 (1988).
- G.F. Adams and C.F. Chabalowski, J. Phys. Chem. 98, 5878 (1994).
- I. Last and T.F. George, J. Chem. Phys. 87, 1183 (1987).
- A.A. Vlasenko, I.S. Lakoba, S.P. Chernov, and P.B. Essel’bakh, Sov. Phys. Dokl. 31, 554 (1986).
- T. Möller, M. Beland, and G. Zimmerer, Chem. Phys. Lett. 136, 551 (1987).
- R.H. Lipson, Chem. Phys. Lett. 129, 82 (1986).
- M. Hamdan, N.W. Copp, D.P. Wareing, J.D.C. Jones, K. Birkinshaw, and N.D. Twiddy, Chem. Phys. Lett. 89, 63 (1982).
- P.J. Hay and T.H. Dunning Jr, J. Chem. Phys. 69, 2209 (1978).
- J.H. Kolts, J.E. Velazco, and D.W. Setser, J. Chem. Phys. 71, 1247 (1979).
- D. Lo and C.E. Zheng, J. Phys. D : Appl. Phys. 20, 714 (1987).
- S.L. Shostak and R.L. Strong, Chem. Phys. Lett. 63, 370 (1979).
- R. Shuker, Appl. Phys. Lett. 29, 785 (1976).
- B.S. Ault and L. Andrews, J. Chem. Phys. 65, 4192 (1976).
- J. Tellinghuisen, J.M. Hoffman, G.C. Tisone, and A.K. Hays, J. Chem. Phys. 64, 2484 (1976).
- J.J. Ewing and C.A. Brau, Phys. Rev. A12, 129 (1975).
- W.Y. Lee, Z.M. Xia, and E.A. Ballik, Mol. Phys. 82, 165 (1994).
- D.W. Setser, H.C. Brashears, and T.D. Dreiling, J. Phys. Colloq. 41, C3-195 (1980).
- P.S. Julienne and M. Krauss, Appl. Phys. Lett. 35, 55 (1979).
- C. Jouvet, C. Lardeux-Dedonder, and D. Solgadi, Chem. Phys. Lett. 156, 569 (1989).
- G. Inoue, J.K. Ku, and D.W. Setser, J. Chem. Phys. 80, 6006 (1984).
- E. Quiñones, Y.C. Yu, D.W. Setser, and G. Lo, J. Chem. Phys. 93, 333 (1990).
- M.A. Goetschalekx, R.L. Mowery, E.R. Krausz, W.C. Yeakel, P.N. Schatz, B.S. Ault, and L. Andrews, Chem. Phys. Lett. 47, 23 (1977).
- J. Tellinghuisen and M.R. Mc Keever, Chem. Phys. Lett. 72, 94 (1980).
- J. Bokor and C.DK. Rhodes, J. Chem. Phys. 73, 2626 (1980).
- H.C. Brashears, Jr. and D.W. Setser, J. Phys. Chem. 84, 224 (1980).
- H.P. Grieneisen, H. Xue-Jing, and K.L. Kompa, Chem. Phys. Lett. 82, 421 (1981).
- R.S.F. Chang, J. Chem. Phys. 76, 2943 (1982).
- Y.C. Yu, D.W. Setser, and H. Horiguchi, J. Phys. Chem. 87, 2199 (1983).
- D.C. Lorents, Proc. International Conference on Lasers’80, San Francisco, California, November 26-30, (1984), p. 575.
- J. Le Calvé, M.C. Castex, B. Jordan, G. Zimmerer, T. Möller, and D. Haaks, Photophysics and Phochemistry Above 6eV, edited by F. Lahmani (Elsevier, Amsterdam, 1985), pp. 639-651.
- M.E. Fajardo and V.A. Apkarian, J. Chem. Phys. 85, 5660 (1986).
- M. Krauss, J. Chem. Phys. 67, 1712 (1977).
- K. Tamagake, J.H. Kolts, and D.W. Setser, J. Chem. Phys. 71, 1264 (1979).
- I.S. Fletcher and D. Husain, J. Chem. Soc. Farad. Trans. II 74, 203 (1978).
- R. Böhling, J. Langen, and U. Schurath, J. Mol. Struct. 222, 171 (1990).
- C.H. Becker, J.J. Valentini, P. Casavecchia, S.J. Sibener, and Y.T. Lee, Chem. Phys. Lett. 61, 1 (1979).
- P. Huxley, D.B. Knowles, J.N. Murrell, and J.D. Watts, J. Chem. Soc. Faraday Trans. 2 80, 1349 (1984).
- V. Aquilanti, D. Cappelletti, V. Lorent, E. Luzzatti, and F. Pirani, Chem. Phys. Lett. 192, 153 (1992).
- A.W. Mc Cown and J.G. Eden, J. Chem. Phys. 81, 2933 (1984).
- A. Sur, A.K. Hui, and J. Tellinghuisen, J. Mol. Spectrosc. 74, 465 (1979).
- S. Szatmari and F.P. Schäfer, Chem. Phys. Lett. 137, 1 (1987).
- J. Tellinghuisen, J. Chem. Phys. 78, 2374 (1983).
- H. Haberland, Z. Phys. A-Atoms and Nuclei 307, 35 (1982).
- K. Johnson, J.P. Simono, P.A. Smith, C. Washington, and A. Kvaran, Mol. Phys. 57, 255 (1986).
- F.J. Adrian and A.N. Jette, J. Chem. Phys. 68, 4696 (1978).
- M.J. Clugston and R.G. Gordon, J. Chem. Phys. 66, 239 (1977).
- K.P. Huber and G. Herzberg, "Molecular Spectra and Molecular Structure", vol. 4. Constants of diatomic molecules (Van Nostrand Reinhold, New York, 1979).
- C.A. Brau and J.J. Ewing, J. Chem. Phys. 63, 4640 (1975).
- A. Kvaran, M.J. Shaw, and J.P. Simons, Appl. Phys. B46, 95 (1988).
- J. Le Calvé and P. Gürtler, J. Chim. Phys. 86, 1847 (1989).
- M.F. Golde, J. Mol. Spectrosc. 58, 261 (1975).
- Q.H. Lou, Hyperfine Interac. 38, 531 (1987).
- N.G. Basov, I.S. Gorban’, V.A. Danilychev, N.G. Zubrilin, and M.P. Chernomorets, Sov. Phys. Dokl. 30, 223 (1985).
- E.E. Muschlitz, Jr., Science 159, 599 (1968).
- D.W. Setser, T.D. Dreiling, H.C. Brashears, Jr., and J.H. Kolts, Farad. Disc. Chem. Soc. 67, 255 (1979).
- C.T. Rettner and J.P. Simons, Farad. Disc. Chem. Soc. 67, 329 (1979).
- A.M. Kosmas, Nuov. Cim. 3D, 981 (1984).
- G. Inoue, J.K. Ku, and D.W. Setser, J. Chem. Phys. 76, 733 (1982).
- S.B. Hassal and E.A. Ballik, J. Appl. Phys. 70, 1042 (1991).
- I.N. Konovalov, V.F. Losev, V.V. Ryzhov, V.F. Tarasenko, and A.G. Tastremskii, Opt. Spectrosc. 47, 137 (1979).
- D.W. Setser and J. Ku, Photophysics and Photochemistry above 6eV, edited by F. Lahmani (Elsevier, Amsterdam, 1985), p. 621-637.
- B.E. Wilcomb and R. Burnham, J. Chem. Phys. 74, 6784 (1981).
- M. Boivineau, J. Le Calvé, M.C. Castex, and C. Jouvet, Chem. Phys. Lett. 130, 208 (1986).
- J.K. Ku, G. Inoue, and D.W. Setser, J. Phys. Chem. 87, 2989 (1983).
- V.S. Pavlenko, S.E. Nalivaïko, V.G. Egorov, O.V. Rzhevskii, and E.B. Gordon, Quantum Electron. 24, 199 (1994).
- J.P. Simons, Chem. Phys. Lett. 91, 484 (1982).
- N.K. Bibinov and I.P. Vinogradov, Sov. J. Chem. Phys. 2, 2693 (1985).
- J.E. Velazco, J.H. Kolts, and D.W. Setser, J. Chem. Phys. 65, 3468 (1976).
- M. Maeda, T. Nishitarumizu, and Y. Miyazoe, Jpn. J. Appl. Phys. 18, 439 (1979).
- M.R. Berman, Chem. Phys. Lett. 157, 562 (1989).
- M.R. Bruce, W.B. Layne, E. Meyer, and J.W. Keto, J. Chem. Phys. 92, 420 (1990).
- J.K. Ku and D.W. Setser, Appl. Phys. Lett. 48, 689 (1986).
- X. Chen and D.W. Setser, J. Phys. Chem. 95, 8473 (1991).
- D.J. Wren, D.W. Setser, and J. Ku, J. Phys. Chem. 86, 284 (1982).
- L.A. Levin, S.E. Moody, E.L. Klosterman, R.E. Center, and J.J. Ewing, IEEE J. Quantum Electron. QE-17, 2282 (1981).
- D. Lin, Y.C. Yu, and D.W. Setser, J. Chem. Phys. 81, 5830 (1984).
- F. Kannari, A. Suda, M. Obara, and T. Fujioka, Appl. Phys. Lett. 42, 766 (1983).
- T. Ishihara and S.C. Lin, Appl. Phys. B48, 315 (1989).
- T. Letardi, H. Fang, and S. Fu, IEEE J. Quantum Electron. QE-28, 1647 (1992).
- F. Kannari, W.D. Kimura, and J.J. Ewing, J. Appl. Phys. 68, 2615 (1990).
- J. Xu, A.R. Slagle, D.W. Setser, and J.C. Ferrero, Chem. Phys. Lett. 137, 63 (1987).
- M. Castillejo, J.M. Figuera, I. Garcia-Moreno, and J.J. Medina, Laser Chem. 12, 13 (1992).
- R.F. Stebbings, F.B. Dunning, and C. Higgs, J. Electr. Spectrosc. Rel. Phen. 23, 333 (1981).
- A.V. Dem’yanov, S.V. Egorov, I.V. Kochetov, A.P. Napartovich, A.A. Pastor, N.P. Penkin, P.Y. Serdobinstev, and N.N. Shubin, Sov. J. Quantum 16, 817 (1986).
- T. Hammer and W. Bötticher, Appl. Phys. B48, 73 (1989).
- C. Gorse, M. Capitelli, S. Longo, E. Estocq, and J. Bretagne, J. Phys. D : Appl. Phys. 24, 1947 (1991).
- S. Longo, M. Capitelli, C. Gorse, A.V. Dem’yanov, I.V. Kochetov, and A.P. Napartovich, Appl. Phys. B54, 239 (1992).
- M. Castillejo, J.M. Figuera, and M. Martin, Chem. Phys. Lett. 117, 181 (1985).
- M.M. Turner and P.W. Smith, IEEE Trans. Plasma Sci. 19, 350 (1991).
- V.S. Zuev, A.V. Kanaev, and L.D. Mikheev, Sov. J. Quantum Electron. 14, 242 (1984).
- M.W. Wilson, M. Rothschild, and C.K. Rhodes, J. Chem. Phys. 78, 3779 (1983).
- T. Ishiwata, A. Tokunaga, and I. Tanaka, Chem. Phys. Lett. 112, 356 (1984).
- R.S. Taylor, Appl. Phys. B41, 1 (1986).
- T.H. Johnson, H.E. Cartland, T.C. Genoni, and A.M. Hunter, J. Appl. Phys. 66, 5707 (1989).
- M.R. Bruce, W.B. Layne, and J.W. Keto, J. Chem. Phys. 92, 428 (1990).
- V.I. Donin and Y.I. Khapov, Sov. J. Quantum Electron. 16, 1034 (1986).
- H.C. Brashears, D.W. Setser, and Y.C. Yu, J. Phys. Chem. 84, 2495 (1980).
- A.K. Shuaibov and V.S. Shevera, Opt. Spectrosc. 47, 224 (1979).
- S.P. Mezyk, R. Cooper, and J. Sherwell, J. Phys. Chem. 95, 3152 (1991).
- M. Tsuji, M. Furusawa, H. Kouno, and Y. Nishimura, J. Chem. Phys. 94, 4291 (1991).
- F. Kannari, A. Suda, M. Obara, and T. Fujioka, IEEE J. Quantum Electron. QE-19, 1587 (1983).
- Z. Ujda, L. Pokora, and M. Stefański, J. Tech. Phys. 32, 387 (1991).
- V.E. Peét and A.B. Treshchalov, Sov. J. Quantum Electron. 15, 1613 (1986).
- M.J. Church and D. Smith, J. Phys. D : Appl. Phys. 11, 2199 (1978).
- G. Imada, H. Nakamura, K. Masugata, W. Masuda, and K. Yatsui, Bull. Nagaoka Univ. Technol. 14, 7 (1992).
- D.R. Bates and W.L. Morgan, Phys. Rev. Lett. 64, 2258 (1990).
- G. Imada, K. Masugata, K. Yatsui, and W. Masuda, Appl. Phys. Lett. 63, 1313 (1993).
- E.P. Glotov, V.A. Danilychev, A.I. Milanich, and A.M. Soroka, Sov. J. Quantum Electron. 9, 1176 (1980).
- A.B. Treshchalov, V.E. Peet, and V.T. Mihkelsoo, IEEE J. Quantum Electron. QE-22, 51 (1986).
- V.F. Losev, V.F. Tarasenko, and Y.I. Bychkov, Sov. J. Quantum Electron. 9, 918 (1979).
- V.M. Baginskii, P.M. Golovinskii, A.M. Razhev, and A.I. Shchedrin, Sov. J. Quantum Electron. 18, 1444 (1988).
- A.A. Alekhin, V.A. Barinov, Y.V. Geras’ko, O.F. Kostenko, F.N. Lyubchenko, and A.V. Tyukavkin, Tech. Phys. 38, 80 (1993).
- H. Furuhashi, M. Ichikawa, E. Fuwa, and T. Goto, IEEE J. Quantum Electron. 29, 1520 (1993).
- G.C. Tysone and J.M. Hoffman, IEEE J. Quantum Electron. QE-18, 1008 (1982).
- M. Ohwa and M.J. Kushner, J. Appl. Phys. 65, 4138 (1989).
- M. Tsuji, T. Muraoka, H. Kouno, and Y. Nishimura, J. Chem. Phys. 97, 1079 (1992).
- W.J. Stevens et M. Krauss, Appl. Phys. Lett. 41, 301 (1982).
- I.V. Chaltakov and I.V. Tomov, Bulg. J. Phys. 15, 70 (1988).
- R.S. Taylor, K.E. Leopold, and K.O. Tan, Appl. Phys. Lett. 59, 525 (1991).
- S.C. Lin, Q.H. Lou, and Q.S. He, J. Quant. Spectrosc. Radiat. Transfer 33, 133 (1985).
- C.H. Fisher, 32 rd Ann. Gaseous Electron. Conf., Pittsburgh, PA, Oct. 1979.
- T.D. Dreiling and D.W. Setser, J. Chem. Phys. 75, 4360 (1981).
- T.G. Finn, R.S.F. Chang, L.J. Palumbo, and L.F. Champagne, Appl. Phys. Lett. 36, 789 (1980).
- K.Y. Tang, D.C. Lorents, R.L. Sharpless, D.L. Huestis, D. Helms, M. Durett, and G.K. Walters, 33rd Gaxous Electronics Conference, Norman, Oklahoma (8 october 1980).
- l. Qihong, Hyperfine Interac. 37, 275 (1987).
- Y.C. Yu, S.J. Wategaonkar, and D.W. Setser, J. Chem. Phys. 96, 8914 (1992).
- H. Hokazono, K. Midorikawa, M. Obara, and T. Fujioka, J. Appl. Phys. 56, 680 (1984).
- M. Maeda, A. Takahashi, T. Mizunami, and Y. Miyazoe, Jpn. J. Appl. Phys. 21, 1161 (1982).
- V.M. Baginskii, P.M. Golovinskii, and A.I. Shchedrin, Sov. Phys. Tech. Phys. 31, 1402 (1986).
- Q. Lou, Opt. Commun. 65, 26 (1988).
- P.K. Miidla, V.E. Peet, R.A. Sorkina, E.E. Tamme, A.B. Treshchalov, and A.V. Sherman, Sov. J. Quantum Electron. 16, 1438 (1986).
- V. Mihkelsoo, P. Miidla, V. Peet, A. Sherman, R. Sorkina, E. Tamme, and A. Treshchalov, J. Phys. B : At. Mol. Opt. 22, 1489 (1989).
- V.M. Baginskii, P.M. Golovinskii, V.A. Danilychev, A.I. Milanich, A.S. Soroka, and A.I. Shchedrin, Sov. J. Quantum Electron. 16, 488 (1986).
- G.P. Glass, F.K. Tittel, W.L. Wilson, M.S. Smayling, and G. Marowsky, Chem. Phys. Lett. 83, 585 (1981).
- T. Mizunami, M. Maeda, O. Uchino, O. Shimomura, and Y. Miyazoe, Rev. Laser Eng. 9, 512 (1981).
- H.C. Brashears, Jr., D.W. Setser, and Y.C. Yu, J. Chem. Phys. 74, 10 (1981).
- B. Forestier, B. Fontaine, and P. Gross, J. Phys. Colloq. 41, C9-455 (1980).
- P.K. Corkum and R.S. Taylor, IEEE J. Quantum. QE-18, 1962 (1982).
- Z.M. Xia and E.A. Ballik, Opt. Commun. 98, 172 (1993).
- R. Tennant, Laser Focus 17, 65 (1981).
- M. Boivineau, J. Le Calvé, M.C. Castex, and C. Jouvet, Chem. Phys. Lett. 128, 528 (1986).
- Y.A. Kudryavtsev and N.P. Kuz’mina, Sov. J. Quantum Electron. 7, 131 (1977).
- O.L. Bourne and A.J. Alcock, Appl. Phys. B32, 193 (1983).
- D.L. Huestis and N.E. Schlotter, J. Chem. Phys. 69, 3100 (1978).
- D.L. Huestis, G. Morowsky, and F.K. Tittel, AIP Conf. Proc. 100, 238 (1983).
- G. Marowsky, F.K. Tittel, W.L. Wilson, Jr., and R. Sauerbrey, AIP Conf. Proc. 100, 334 (1983).
- W.L. Morgan and D.R. Bates, J. Phys. B : At. Mol. Opt. Phys. 25, 5421 (1992).
- A.W. Mc Cown, M.N. Ediger, S.M. Stazak, and J.G. Eden, AIP Conf. Proc. 100, 222 (1983).
- M. Ohwa and M. Obara, J. Appl. Phys. 59, 32 (1986).
- M.E. Fajardo and V.A. Apkarian, J. Chem. Phys. 89, 4102 (1988).
- J.G. Mc Caffrey, H. Kunz, and N. Schwentner, J. Chem. Phys. 96, 2825 (1992).
- H. Jara, M. Shahidi, H. Pummer, H. Egger, and C.K. Rhodes, AIP Conf. Proc. 146, 132 (1986).
- I. Last and T.F. George, J. Chem. Phys. 86, 3787 (1987).
- G. Marowsky, R. Sauerbrey, F.K. Tittel, and W.L. Wilson, Jr., Chem. Phys. Lett. 98, 167 (1983).
- F. Okada and V.A. Apkarian, J. Chem. Phys. 94, 133 (1991).
- K.Y. Tang and D.C. Lorents, in Proceedings of the International Conference on Lasers’81 (STS, Mc Lean, VA, 1981).
- W.S. Zuev, A.V. Kanaev, and L.D. Mikheev, Sov. J. Quantum Electron. 17, 884 (1987).
- V.S. Dubov and Y.E. Lapsker, Sov. J. Quantum Electron. 13, 1240 (1983).