JUNQ et IPOD
Les JUNQ et IPOD constituent des types de structures d'inclusion de protéines cytosoliques qu'on retrouve chez les eucaryotes.
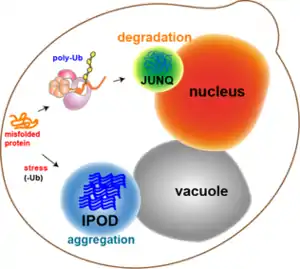
Des maladies neurodégénératives, comme celles de Parkinson, Alzheimer et Huntington, sont associées et corrélées à l'agrégation des protéines et l'accumulation de protéines mal repliées dans ces structures d'inclusion. Pendant de nombreuses années, l'agrégation protéique était considérée comme un processus aléatoire par lequel les protéines mal repliées se collent entre elles pour former des inclusions[1] (s'imaginer un paquet de cheveux qui s'empilent de façon désordonnée dans le coin d'une pièce). De plus, on pensait que les agrégats protéiques étaient des agents toxiques ainsi que la cause d'un dysfonctionnement neuronal pouvant conduire à la mort. Cependant, des études récentes utilisant des méthodes modernes (comme la microscopie à fluorescence) ont montré que l'agrégation protéique peut en fait être un processus fermement régulé et organisé par lequel la cellule se protège des protéines toxiques par séquestration des structures d'inclusion[2]. En 2008, Daniel Kaganovich a montré que les cellules eucaryotes trient les protéines mal repliées et les transportent vers deux structures d'inclusion distinctes via un processus cellulaire bien rodé[3], qui sont :
- Le JUNQ (pour JUxta Nuclear Quality control compartment en anglais, soit compartiment de contrôle qualité juxta-nucléaire en français)
- L'IPOD (pour Insoluble Protein Deposit en anglais, soit dépôt pour les protéines insolubles en français)
Les JUNQ et IPOD sont évolutivement conservés et se trouvent dans des sites cellulaires spécifiques et définis. La distribution des protéines mal repliées et agrégées au JUNQ et IPOD requiert un cytosquelette intact et des composants de contrôle qualité cellulaires spécifiques, comme les protéines de choc thermique (HSP en anglais)[4]. La division en deux structures d'inclusion distinctes sont dues aux différents traitements et gestions des différentes sortes de protéines mal repliées (par exemple les protéines ubiquitinées versus non ubiquitinées). La ségrégation des agrégats protéiques toxiques vers les structures d'inclusion JUNQ et IPOD est un moyen par lequel les cellules mammaliennes peuvent être rajeunies par division asymétrique[5].
Ainsi, la découverte des JUNQ et IPOD a fourni une nouvelle perspective saisissante de comment les cellules prennent en charge les protéines agrégées et a donné une preuve convaincante que l'agrégation protéique est un processus cellulaire non aléatoire, bien régulé et bien contrôlé. De plus, la découverte de JUNQ et IPOD a suggéré qu'en plus d'un contrôle qualité temporaire (c'est-à-dire dont la durée dépend de la gestion des protéines endommagées), les cellules exploitent l'homéostasie de façon spatiale[5]. Si la dégradation n'est pas possible, la protection de l'environnement cellulaire contre une protéine mal repliée est accomplie par la séquestration de celle-ci dans une structure d'inclusion pour agrégats.
Contexte
Afin de fonctionner correctement, la majorité des protéines doit préserver une structure tridimensionnelle de faible énergie, connue sous le nom d'état natif. La stabilité d'une protéine est fermement régulée au long des étapes de sa vie : depuis sa naissance (synthèse au niveau du ribosome), par l'assemblage ou au repliement, jusqu'à sa mort – lorsque la protéine est dégradée et quitte l'environnement cellulaire[6]. L'homéostasie protéique (protéostasie)[7] résulte de l'action coordonnée des différents "bras" du système de contrôle qualité cellulaire : chaperones moléculaires, protéases et autres facteurs de régulation. En conséquence, la viabilité cellulaire dépend d'une gestion efficace et dans les délais des protéines mal repliées. Une telle gestion par la machinerie de contrôle qualité inclut la reconnaissance des protéines mal repliées par les chaperones et les ubiquitine ligases, l'ubiquitination et la dégradation.
L'effondrement de la protéostasie, due à l'endommagement, le stress, les mutations et la vieillesse, s'est avéré être la base d'un grand nombre de maladies humaines communes, telles que les maladies neurodégénératives[8]. Bien que causées par différentes sortes de protéines mutées (par exemple la protéine Huntingtine dans la maladie de Huntington) et perturbant des tissus distincts (par exemple le striatum dans la maladie de Huntington), de telles maladies partagent une caractéristique commune : l'accumulation de protéines mal repliées dans les structures d'inclusion. Ainsi, on a pensé que ces structures d'inclusion étaient la cause de telles maladies. Cependant, la nature et les caractéristiques de ces structures d'inclusion intracellulaires échappaient aux chercheurs. Plusieurs sortes de protéines (comme les prions ou les substrats de dégradation des protéines associée au réticulum endoplasmique) ont été soupçonnées de former différentes sortes de structures d'inclusion (par exemple les agrésomes ou les amyloïdes) ; malgré tout on ne sait pas si ces observations se combinent en une seule et sont liées au même site sub-cellulaire. De plus, les voies conduisant à la formation de structures d'inclusion et l'implication de la machinerie cellulaire de contrôle qualité protéique étaient indéfinies et inconnues. Ainsi, il était nécessaire de faire une étude systématique fournissant une compréhension globale de l'agrégation protéique et des structures d'inclusion. La découverte de JUNQ et IPOD[3] a donné de nouveaux aperçus de comment la cellule prend en charge différentes sortes de protéines mal repliées et a offert un cadre de travail inédit pour assembler le grand "puzzle" de l'agrégation protéique[2].
Découverte
Le sort des protéines mal repliées et le processus menant à la formation d'inclusions d'agrégats ont été en premier lieu étudiés en utilisant des méthodes biochimiques (par exemple le Western blot).
Un aperçu plus approfondi du processus biologique du contrôle qualité des protéines et de l'agrégation a été possible par une approche innovante du problème, appelée "Imagerie des Cellules Vivantes" ("Live Cell Imaging" en anglais)[9].
L'ICV permet de suivre les protéines in vivo dans l'espace et le temps, au sein de leur environnement naturel endogène. Ainsi, une telle méthode fournit plus d'informations sur la dynamique et les étapes d'événements et processus biologiques. La méthode tire profit de la facilité de détecter des protéines fluorescentes fusionnées à une protéine d'intérêt, qui peut être ensuite suivie à l'intérieur d'une cellule en utilisant un microscope à fluorescence. La cellule peut ensuite être soumise à une perturbation d'intérêt (par exemple un médicament, l'expression d'une protéine mal repliée...), et plusieurs propriétés de la protéine tagguée pour fluorescer peuvent être analysées en utilisant la vidéomicroscopie :
- Changements dans le niveau de fluorescence, indiquant des changements de niveaux d'expression (des niveaux plus élevés correspondent à une régulation positive d'une protéine)
- Changements de localisation (par exemple migration d'une protéine du cytosol vers le noyau)
- Solubilité (par exemple en utilisant la FRAP[10])
- Interaction avec l'environnement intracellulaire (par exemple en utilisant la FLIP[10])
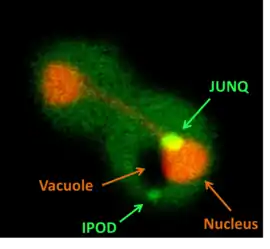
Afin de surveiller le sort des protéines cytosoliques mal repliées in vivo, un plasmide portant un rapporteur de repliement marqué à la GFP a été cloné. Le rapporteur de repliement, constitué d'une protéine modèle pour l'agrégation, était un Ubc9 (enzyme SUMO) mutant (UBC9ts), abritant une mutation faux-sens (Y68L) avec un phénotype sensible à la température (ts)[11] - [12]. La Ubc9ts légèrement stable est pleinement fonctionnelle en conditions physiologiques permissives (25 °C) grâce aux chaperones cellulaires actives. Le complexe GFP-Ubc9ts a subi une transformation dans des levures et a été visualisée en utilisant un microscope à fluorescence.
On a pensé que surveiller le détecteur de repliement GFP-Ubc9ts permettait d'indiquer la protéostasie cellulaire et d'analyser la capacité du système cellulaire de contrôle qualité protéique afin prendre en charge plusieurs sortes de stress. On a ensuite observé qu'en conditions normales, le complexe GFP-Ubc9ts est diffusé dans le noyau et le cytosol. Cependant, en conditions de choc thermique, le complexe a formé des structures cytosoliques ponctuées. De manière remarquable, lorsque le protéasome a été altéré et l'enlèvement de la protéine mal repliée par la dégradation a été bloquée, on a observé la formation de deux inclusions cytosoliques distinctes. Des méthodes biochimiques standard et conservatives, telles la fractionation cellulaire et le Western blot, n'auraient pas révélé la partition en deux types d'agrégats cytosoliques.
Les deux inclusions détectées se sont avérées être des compartiments de contrôle qualité évolutivement conservés, avec des caractéristiques différentes et des fonctions distinctes. Ils ont été appelés JUNQ et IPOD[3] et représentent deux voies cellulaires pour la séquestration et la prise en charge des protéines potentiellement toxiques et enclines à l'agrégation.
La séparation des substrats de contrôle qualité (c'est-à-dire les protéines mal repliées) vers l'un ou l'autre des compartiments dépend de leur statut d'ubiquitination et leur état d'agrégation (c'est-à-dire leur solubilité).
Les protéines ubiquitinées sont transportées au JUNQ, où elles sont traitées pour être dégradées par le protéasome. Les protéines mal repliées qui ne sont pas ubiquitinées et agrégées in fine sont séquestrées dans l'IPOD.
Ainsi, la location sub-cellulaire d'une protéine mal repliée (c'est-à-dire dans le JUNQ ou l'IPOD) fournit des informations sur son inertaction avec la machinerie cellulaire de contrôle qualité protéique (par exemple sa ligase E3).
Le JUNQ
_tethered_to_the_nucleus_(orange).tif.jpg.webp)
Importance
Afin de maintenir l'homéostasie cellulaire, le système cellulaire de contrôle qualité doit distinguer les protéines bien repliées et mal repliées. Une protéine mal repliée peut être reconnue et fermement prise en charge soit par un néo-repliement, soit par son ubiquitination et sa dégradation par le protéasome.
Cependant, l'augmentation cellulaire des charges protéiques mal repliées, due à divers types de stress (choc thermique...), peut causer une saturation et épuiser la machinerie de contrôle qualité. Dans de tels cas, la dégradation des protéines mal repliées n'est pas possible, et une seconde ligne de mécanisme de défense cellulaire active doit être exécutée : diriger les protéines mal repliées vers des sites cellulaires spécifiques[2].
Le JUNQ sert dans ce cas de site de séquestration. Il a été montré[3] que lorsque le protéasome est perturbé (par exemple par des faibles niveaux d'expression de la sous-unité du protéasome RPN11), les protéines mal repliées ubiquitinées sont rangées dans le JUNQ. Après récupération à la suite de conditions de stress (par exemple d'un choc thermique à une température permissive), les protéines mal repliées qui s'accumulent dans le JUNQ peuvent être soit néo-repliées par la machinerie cellulaire chaperone, ou dégradées par le protéasome 26S. Ainsi, la séquestration d'une protéine dans le JUNQ est réversible.
Propriétés
Le JUNQ est un site cellulaire non lié à la membrane et localisé au bord du noyau, à forte proximité du réticulum endoplasmique. Des analyses FRAP et FLIP ont révélé que les protéines du JUNQ sont solubles et s'échangent entre le JUNQ et le cytosol, suggérant que le JUNQ a une structure dynamique.
Le transport au JUNQ dépend des chaperones moléculaires et des co-chaperones, ainsi que du cytosquelette d'actine[4]. Les protéines mal repliées doivent être ubiquitinées afin d'être envoyées au JUNQ. Si l'ubiquitination est bloquée, une protéine mal repliée sera dirigée vers l'IPOD. L'accumulation de protéines mal repliées déclenche le recrutement des protéasomes 26S dans le JUNQ.
L'IPOD
_tethered_to_the_vacuole_(green).tif.jpg.webp)
Importance
Il devient de plus en plus évident que la capacité cellulaire à maintenir la protéostasie[7] décline avec l'âge[8], causant ainsi la manifestation tardive de maladies neurodégénératives. Dans de telles maladies (Huntington par exemple), une protéine mutée se replie mal et devient toxique pour l'environnement cellulaire de plusieurs façons différentes, comme sous forme de protéines cytosoliques dénaturantes[13]. Incapable de dégrader ces espèces toxiques, la cellule doit les isoler afin d'éviter leur interaction dangereuse avec le protéome cellulaire. On a montré que l'IPOD[3] était le site sub-cellulaire dans lequel les protéines amyloïdogéniques sont séquestrées, servant ainsi de compartiment de contrôle qualité protecteur.
De plus, il a été suggéré par le groupe de Susan Lindquist que l'IPOD est le site où les prions de levures subissent un processus de maturation[14]. Ainsi, l'IPOD peut servir non seulement de site de séquestration, mais aussi de compartiment fonctionnel[14].
Propriétés
L'IPOD est une site cellulaire non lié à la membrane, localisé chez les levures dans la vacuole. Des analyses FRAP et FLIP ont révélé que les protéines dans l'IPOD sont solidement empaquetées, insolubles et ne subissent pas d'échanges avec le cytosol. Les protéines amyloïdogéniques, telles que la Huntingtine, constituent les subtrats de l'IPOD.
Les protéines mal repliées doivent être non ubiquitinées afin d'être dirigées vers l'IPOD. L'ubiquitination d'un substrat autre que de l'IPOD, tel que le prion fongique RNQ1, résultera en sa séquestration dans le JUNQ.
Lors de l'accumulation de protéines mal repliées, la chaperone désagrégée AAA HSP104, se localise dans l'IPOD. Il reste pourtant à déterminer si la HSP104 fonctionne dans l'IPOD ou est simplement séquestrée là en étant accrochée à un substrat.
La structure pré-autophagosomale (SAP) est localisée par l'IPOD[15]. Cependant, il n'a pas été montré que les substrats d'IPOD sont transportées vers la vacuole, et ainsi le lien entre l'IPOD et l'autophagie reste encore à déterminer[3].
Voir aussi
Notes
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « JUNQ and IPOD » (voir la liste des auteurs).
Articles connexes
Références
- Sebastian Treusch, Douglas M. Cyr et Susan Lindquist, « Amyloid deposits: protection against toxic protein species? », Cell Cycle (Georgetown, Tex.), vol. 8, no 11, , p. 1668–1674 (ISSN 1551-4005, PMID 19411847, PMCID PMC4451085, DOI 10.4161/cc.8.11.8503, lire en ligne, consulté le )
- Jens Tyedmers, Axel Mogk et Bernd Bukau, « Cellular strategies for controlling protein aggregation », Nature Reviews. Molecular Cell Biology, vol. 11, no 11, , p. 777–788 (ISSN 1471-0080, PMID 20944667, DOI 10.1038/nrm2993, lire en ligne, consulté le )
- Daniel Kaganovich, Ron Kopito et Judith Frydman, « Misfolded proteins partition between two distinct quality control compartments », Nature, vol. 454, no 7208, , p. 1088–1095 (ISSN 1476-4687, PMID 18756251, PMCID PMC2746971, DOI 10.1038/nature07195, lire en ligne, consulté le )
- Sebastian Specht, Stephanie B. M. Miller, Axel Mogk et Bernd Bukau, « Hsp42 is required for sequestration of protein aggregates into deposition sites in Saccharomyces cerevisiae », The Journal of Cell Biology, vol. 195, no 4, , p. 617–629 (ISSN 1540-8140, PMID 22065637, PMCID PMC3257523, DOI 10.1083/jcb.201106037, lire en ligne, consulté le )
- Mikołaj Ogrodnik, Hanna Salmonowicz, Rachel Brown et Joanna Turkowska, « Dynamic JUNQ inclusion bodies are asymmetrically inherited in mammalian cell lines through the asymmetric partitioning of vimentin », Proceedings of the National Academy of Sciences of the United States of America, vol. 111, no 22, , p. 8049–8054 (ISSN 1091-6490, PMID 24843142, PMCID PMC4050583, DOI 10.1073/pnas.1324035111, lire en ligne, consulté le )
- Richard I. Morimoto et Ana M. Cuervo, « Protein homeostasis and aging: taking care of proteins from the cradle to the grave », The Journals of Gerontology. Series A, Biological Sciences and Medical Sciences, vol. 64, no 2, , p. 167–170 (ISSN 1758-535X, PMID 19228787, PMCID PMC2655025, DOI 10.1093/gerona/gln071, lire en ligne, consulté le )
- Evan T. Powers, Richard I. Morimoto, Andrew Dillin et Jeffery W. Kelly, « Biological and chemical approaches to diseases of proteostasis deficiency », Annual Review of Biochemistry, vol. 78, , p. 959–991 (ISSN 1545-4509, PMID 19298183, DOI 10.1146/annurev.biochem.052308.114844, lire en ligne, consulté le )
- Anat Ben-Zvi, Elizabeth A. Miller et Richard I. Morimoto, « Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging », Proceedings of the National Academy of Sciences of the United States of America, vol. 106, no 35, , p. 14914–14919 (ISSN 1091-6490, PMID 19706382, PMCID PMC2736453, DOI 10.1073/pnas.0902882106, lire en ligne, consulté le )
- « Live-Cell Imaging », sur Nikon’s MicroscopyU (consulté le )
- Jennifer Lippincott-Schwartz et George H. Patterson, « Development and use of fluorescent protein markers in living cells », Science (New York, N.Y.), vol. 300, no 5616, , p. 87–91 (ISSN 1095-9203, PMID 12677058, DOI 10.1126/science.1082520, lire en ligne, consulté le )
- J. Betting et W. Seufert, « A yeast Ubc9 mutant protein with temperature-sensitive in vivo function is subject to conditional proteolysis by a ubiquitin- and proteasome-dependent pathway », The Journal of Biological Chemistry, vol. 271, no 42, , p. 25790–25796 (ISSN 0021-9258, PMID 8824207, lire en ligne, consulté le )
- P. Tongaonkar, K. Beck, U. P. Shinde et K. Madura, « Characterization of a temperature-sensitive mutant of a ubiquitin-conjugating enzyme and its use as a heat-inducible degradation signal », Analytical Biochemistry, vol. 272, no 2, , p. 263–269 (ISSN 0003-2697, PMID 10415098, DOI 10.1006/abio.1999.4190, lire en ligne, consulté le )
- Jeremy L. England et Daniel Kaganovich, « Polyglutamine shows a urea-like affinity for unfolded cytosolic protein », FEBS letters, vol. 585, no 2, , p. 381–384 (ISSN 1873-3468, PMID 21176779, DOI 10.1016/j.febslet.2010.12.023, lire en ligne, consulté le )
- Jens Tyedmers, Sebastian Treusch, Jijun Dong et J. Michael McCaffery, « Prion induction involves an ancient system for the sequestration of aggregated proteins and heritable changes in prion fragmentation », Proceedings of the National Academy of Sciences of the United States of America, vol. 107, no 19, , p. 8633–8638 (ISSN 1091-6490, PMID 20421488, PMCID PMC2889312, DOI 10.1073/pnas.1003895107, lire en ligne, consulté le )
- K. Suzuki, T. Kirisako, Y. Kamada et N. Mizushima, « The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation », The EMBO journal, vol. 20, no 21, , p. 5971–5981 (ISSN 0261-4189, PMID 11689437, DOI 10.1093/emboj/20.21.5971, lire en ligne, consulté le )