Perte de fluorescence induite par photoblanchiment
La Perte de fluorescence induite par photoblanchiment, plus communément désignée par son acronyme anglais FLIP (pour Fluorescence Loss Induced by Photobleaching), est une technique de microscopie à fluorescence qu'on utilise pour observer le mouvement de molécules à l'intérieur des cellules et des membranes. En général, on "taggue" une membrane cellulaire avec un colorant fluorescent afin de visualiser ce que l'on veut observer. Une zone spécifique de cette partie tagguée est ensuite "blanchie" plusieurs fois en utilisant le rayon d'un laser d'un microscope confocal. Après chaque balayage du microscope, le blanchiment a lieu à nouveau. Ce processus est répété plusieurs fois afin d'assurer que tous les fluorochromes accessibles soient blanchis, car les fluorochromes non blanchis sont échangés avec les fluorochromes blanchis, ce qui cause un mouvement à travers la membrane cellulaire. La quantité de fluorescence issue de cette région est ensuite mesurée sur une certaine période de temps afin de déterminer les résultats du photoblanchiment de la cellule en entier.
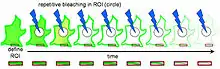
Dispositif expérimental
Avant que le photoblanchiment puisse avoir lieu, on doit injecter une protéine fluorescente dans les cellules (on utilise généralement une GFP), ce qui va permettre aux protéines ciblées d'émettre de la fluorescence et ainsi de pouvoir les suivre au cours du processus. Ensuite, on doit définir une région d'intérêt ; cette région d'intérêt initiale contient généralement la cellule en entier ou bien plusieurs cellules. En FLIP, le photoblanchiment a lieu juste en dehors de cette région d'intérêt, c'est pourquoi une région de photoblanchiment doit également être définie. On doit enfin déterminer une troisième région, celle où la mesure se fera. Un certain nombre de balayages initiaux doit être fait afin de d'identifier la fluorescence avant le photoblanchiment. Ces balayages serviront de contrôles auxquels on comparera les balayages photoblanchis. À la suite de cela, le photoblanchiment peut enfin avoir lieu. Entre chaque "pulse" de blanchiment, il est nécessaire de laisser du temps au matériel fluorescent pour revenir à son état initial. Il est également important de prendre plusieurs balayages de la région d'intérêt immédiatement après chaque pulse de blanchiment pour étude complémentaire approfondie[1]. Le changement du niveau de fluorescence dans la région d'intérêt peut ensuite être quantifié de trois façons différentes. La plus commune est de choisir l'emplacement, la taille et le nombre de régions d'intérêt en se basant sur une inspection visuelle des groupes d'images à disposition. Les deux autres, plus récentes mais plus fiables, consistent soit à détecter des zones de mobilité de sonde différente sur base d'image individuelle, soit à modéliser physiquement la perte de fluorescence des corps en mouvement[2].
La perte de fluorescence est définie par la fraction mobile, ou la fraction de fluorochromes capables de revenir à leur état initial au sein d'une zone photoblanchie, de la protéine tagguée pour fluorescer. Une perte incomplète de fluorescence indique qu'il y a des fluorochromes qui ne bougent pas, ou bien qui transitent vers la zone blanchie. Cela permet d'identifier la fraction immobile, ou la fraction de fluorochromes incapable de revenir à son état initial au sein d'une zone photoblanchie, des protéines tagguées pour fluorescer. L'immobilité indique qu'il y a des protéines pouvant se trouver dans des compartiments isolés du reste de la cellule, les empêchant d'être affectées par le photoblanchiment répété[3].
Applications
Vérifier la continuité des organites à membrane
L'utilisation originale de la FLIP consiste à déterminer la continuité des organites à membrane. Cette continuité (ou absence de celle-ci) est déterminée en observant la quantité de fluorescence dans la région d'intérêt. S'il y a une perte complète de fluorescence, cela indique que les organites sont continus. Cependant, s'il y a une perte incomplète de fluorescence, il n'y a alors pas de continuité entre les organites. Au lieu de cela, ces organites sont alors compartimentés et ainsi isolés du transfert de tous les fluorochromes photoblanchis[3]. La continuité de l'appareil de Golgi, du réticulum endoplasmique et du noyau a été vérifiée en utilisant la FLIP.
Taux d'échange entre le noyau et le cytoplasme
Deux autres utilisations moins courantes de la FLIP visent à déterminer la façon dont les protéines sont transportées du cytoplasme au noyau, et ensuite déterminer le taux auquel ce transport a lieu. Afin de déterminer quelles portions sont impliquées dans ce transport et à quel moment, on observe des balayages continus. Au plus vite une partie du cytoplasme est utilisée dans le processus de transport, au plus vite on y observera une perte complète de fluorescence[4]. L'image résultante de ce processus doit être un cytoplasme complètement photoblanchi. Si la cellule participe aussi à l'export du noyau vers le cytoplasme, un photoblanchiment aura également lieu dans le noyau.
Le taux d'échange entre le noyau et le cytoplasme peut aussi être déterminé à partir de ce type de données. Dans ces exemples, la région de photoblanchiment se trouve dans le noyau. Si le transport survient à un taux rapide, les niveaux de fluorescence au sein des compartiments nucléaires vont rapidement diminuer au niveau des cadres sélectionnés. Cependant, si le transport a lieu à un taux lent, les niveaux de fluorescence vont rester les mêmes ou bien diminuer seulement de façon très légère[4].
FLIP vs FRAP
La FLIP est souvent utilisée en étroite association avec la FRAP (redistribution de fluorescence après photoblanchiment). La différence majeure entre ces deux techniques de microscopie réside dans le fait que la FRAP implique l'étude de la capacité d'une cellule à se remettre d'un seul événement de photoblanchiment alors que la FLIP implique l'étude de comment la perte de fluorescence se répand à travers la cellule après de multiples événements de photoblanchiment. Cette différence d'objectif conduit également à une différence dans les parties de la cellule qu'on observe. En FRAP, la zone qui est vraiment photoblanchie correspond à la zone d'intérêt. À l'inverse, en FLIP, la région d'intérêt est juste à l'extérieur de la région qui est photoblanchie. Une autre différence importante est qu'en FRAP, il n'y a qu'un seul événement de photoblanchiment ainsi qu'une période de récupération afin d'observer à quel point les fluorochromes retournent de façon efficace au site blanchi ; alors qu'en FLIP, des événements multiples de photoblanchiment ont lieu afin d'empêcher le retour des fluorochromes non blanchis à la région de blanchiment. Comme la FLIP, la FRAP est utilisée dans l'étude de la continuité des organites à membrane. Ces deux techniques sont souvent utilisées ensemble afin de déterminer la mobilité des protéines tagguées à la GFP[3]. La FLIP peut aussi être utilisée pour mesurer le transfert moléculaire entre des régions d'une cellule sans tenir compte du taux de mouvement. Cela permet d'avoir une analyse plus poussée du trafic de protéines au sein d'une cellule. Cela diffère de la FRAP qui est principalement utile pour déterminer la mobilité des protéines uniquement dans les régions où a lieu le photoblanchiment[5].
Complications potentielles
Il y a plusieurs complications qu'impliquent à la fois la FLIP et la FRAP. Comme ces deux techniques de microscopie observent des cellules vivantes, il y a toujours la possibilité que les cellules bougent, ce qui fausse les résultats. La meilleure façon d'éviter cela est d'utiliser un algorithme d'alignement, qui va compenser chaque mouvement et éliminer une large partie de l'erreur due au mouvement. Dans ces expériences, il est également capital d'avoir un groupe contrôle afin d'ajuster les résultats et corriger la courbe de récupération pour la perte globale de fluorescence[3]. Une autre façon de minimiser l'erreur est de restreindre le photoblanchiment au niveau d'une seule région ou d'une seule zone. Cette limitation va jouer le rôle de contrôle et va limiter la perte de fluorescence due au photo-dommage par rapport à la perte de fluorescence due au photoblanchiment[3].
Voir aussi
Notes
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Fluorescence loss in photobleaching » (voir la liste des auteurs).
Articles connexes
Références
- (en) Robert C. Dickson et Michael D. Mendenhall, Signal Transduction Protocols (2nd ed.), Springer Science & Business Media, , 327 p. (ISBN 978-1-59259-816-8, lire en ligne), p. 300-304
- Daniel Wüstner, Lukasz M. Solanko, Frederik W. Lund et Daniel Sage, « Quantitative fluorescence loss in photobleaching for analysis of protein transport and aggregation », BMC bioinformatics, vol. 13, , p. 296 (ISSN 1471-2105, PMID 23148417, PMCID PMC3557157, DOI 10.1186/1471-2105-13-296, lire en ligne, consulté le )
- (en) Hellen C. Ishikawa-Ankerhold, Richard Ankerhold et Gregor P. C. Drummen, « Advanced Fluorescence Microscopy Techniques—FRAP, FLIP, FLAP, FRET and FLIM », Molecules, vol. 17, no 4, , p. 4047–4132 (DOI 10.3390/molecules17044047, lire en ligne, consulté le )
- (en) R. G. Davies, D. A. Jans et K. M. Wagstaff, « Use of fluorescence photobleaching techniques to measure the kinetics of intracellular transport », Microscopy: Science, Technology, Applications, and Education, (lire en ligne)
- (en) Akira Kitamura et Hiroshi Kubota, « Amyloid oligomers: dynamics and toxicity in the cytosol and nucleus », FEBS Journal, vol. 277, no 6, , p. 1369–1379 (ISSN 1742-4658, DOI 10.1111/j.1742-4658.2010.07570.x, lire en ligne, consulté le )