Dysplasie ventriculaire droite arythmogène
La dysplasie ventriculaire droite arythmogène (DVDA) est une maladie cardiaque, de type canalopathie, responsable de troubles du rythme ventriculaire pouvant conduire à la mort subite chez les personnes jeunes et les athlètes.
| Dysplasie ventriculaire droite arythmogène | |
| Référence MIM | 107970 |
|---|---|
| Transmission | Dominante |
| Chromosome | Voir article |
| Gène | Voir article |
| Empreinte parentale | Non |
| Mutation | Ponctuelle |
| Mutation de novo | Possible |
| Nombre d'allèles pathologiques | Plus de 300 connues |
| Prévalence | 6/10 000 jusqu'à 4/1 000[1] |
| Pénétrance | 50 % |
| Maladie génétiquement liée | tachycardie ventriculaire polymorphe catécholergique |
| Diagnostic prénatal | Possible |
| Liste des maladies génétiques à gène identifié | |
| Spécialité | Cardiologie |
|---|
| CIM-10 | I42.8 |
|---|---|
| OMIM | 107970 |
| DiseasesDB | 29750 |
| eMedicine | 163856 |
| MeSH | D019571 |
| Patient UK | Arrhythmogenic-right-ventricular-cardiomyopathy |
![]() Mise en garde médicale
Mise en garde médicale
C'est une forme de cardiomyopathie (littéralement, maladie du muscle cardiaque) d'origine non ischémique intéressant prioritairement le ventricule droit.
Cette maladie se caractérise par le remplacement des cellules musculaires du ventricule droit par des cellules adipeuses. L'infiltration graisseuse commence dans le ventricule droit (au niveau de la paroi libre du ventricule) et atteint secondairement le ventricule gauche.
Les manifestations cliniques de cette maladie sont très variables d'un individu à l'autre.
Historique
Giovanni Maria Lancisi décrit dès 1736 une maladie familiale comportant une atteinte du ventricule droit et se compliquant de mort subite[2]. La maladie a été décrite en tant que telle pour la première fois en 1978 par une équipe parisienne, la nommant de son nom actuel[3]. Le terme de « ventricule papyracé » était alors parfois utilisé[4] pour désigner cette atteinte mais n'est plus guère utilisé. Le « crochetage » de l'onde QRS de l'ECG, appelé « onde epsilon », a été décrit en 1984[5].
Épidémiologie
La dysplasie ventriculaire droite arythmogène est principalement rencontrée chez l'homme. Sa prévalence est de l'ordre de 1 cas sur 2 500 à 5 000 personnes. La maladie est présente à travers le monde, mais semble plus prévalente dans les populations du bassin méditerranéen, notamment en Italie du Nord et en Grèce, dû à des facteurs génétiques[6] - [7]. Elle est diagnostiquée essentiellement chez l'adulte jeune[8].
Physiopathologie
Elle est peu connue. L'apoptose (mort cellulaire programmée) semble y jouer un grand rôle[9]. Les raisons pour lesquelles le ventricule droit est prioritairement atteint ne sont pas connues.
Mécanismes physiopathologiques
La dysplasie ventriculaire droite arythmogène semble être due à une atteinte des jonctions intercellulaires au niveau du muscle cardiaque (desmosomes)[8] par l'atteinte d'un ou plusieurs gènes intervenant dans la synthèse de protéines impliquées dans celles-ci. Cela entraînerait le détachement des cellules et leur mort.
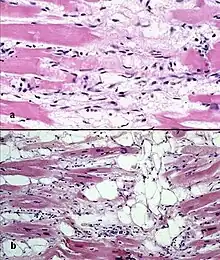
Le processus de transformation du tissu cardiaque en tissu fibro-graisseux démarre dans la région épicardique et progresse vers la surface endocardique. Une dilatation anévrismale est retrouvée dans 50 % des séries autopsiques. Elle survient habituellement dans les régions diaphragmatiques, apicales et infundibulaires.
Le ventricule gauche est atteint dans 50 à 60 % des cas. Si le ventricule gauche est touché, cela signifie que la maladie est déjà bien avancée et de mauvais pronostic.
Il existe deux lésions pathologiques dans la dysplasie ventriculaire droite arythmogène : l'infiltration adipeuse et l'infiltration fibro-adipeuse :
- l'infiltration adipeuse est confinée au ventricule droit. Elle implique une substitution partielle ou quasi complète du myocarde par du tissu adipeux, sans amincissement de la paroi. Le ventricule gauche et le septum inter-ventriculaire (paroi séparant les deux ventricules) sont généralement épargnés. Il n'y a pas d'infiltration inflammatoire. Une dégénérescence des myocytes est mise en évidence dans la moitié des cas ;
- l'infiltration fibro-adipeuse implique le remplacement des myocytes par du tissu fibro-adipeux. Il existe une infiltration inflammatoire (principalement de lymphocytes T). L'atrophie du myocarde est liée aux lésions et à l'apoptose. Ceci conduit à un amincissement de la paroi libre du ventricule droit.
Cette transformation interfère avec la transmission de l'influx électrique, expliquant les anomalies de l'électrocardiogramme et les troubles du rythme, l'atteinte des jonctions inter-cellulaires pouvant également expliquer le tout[10].
Un mécanisme auto-immun est possible puisque des autoanticorps anti-myocyte et anti-disques intercalés sont très souvent retrouvés[11].
Au niveau cellulaire, il existe des lésions de la membrane nucléaire pouvant conduire à une fragilisation de l'ADN, du moins chez ceux porteurs d'une mutation sur la plakophiline 2[12].
Causes
Une origine familiale est retrouvée dans près de la moitié des cas[8]. La transmission s'effectue sur le mode autosomique dominant, avec une expression variable. La pénétrance est d'environ 20 à 35 %.
Les trois gènes connus pour être associés à cette pathologie sont : le RYR2 qui code les récepteurs cardiaques de la ryanodine, le DSP qui code la desmoplakine et le PKP2 qui code la plakophiline 2. Trois autres gènes codent des protéines intervenant dans les jonctions inter-cellulaires : DSG2, DSC2 et JUP. Trois autres gènes ont un rôle plus discuté : TGFβ3(transforming growth factor-beta3)[13], RYR2 (ryanodine receptor)[14] et TMEM43 (transmembrane protein 43)[15]. Des mutations ont été décrites sur quatre autres locus, tous près du gène codant la titine, protéine du muscle cardiaque, ainsi que sur le gène lui-même (TTN)[16]. La proportion de mutation de novo est inconnue.
Il existe une variante autosomique récessive de cette pathologie, décrite initialement sur l'île grecque de Naxos[17]. La pénétrance est supérieure à 90 %. La maladie de Naxos est caractérisée par la triade : dysplasie ventriculaire droite arythmogène, kératose palmoplantaire et cheveux laineux.
Des modèles animaux de la maladie ont été développés, aidant à la compréhension de l'évolution de celle-ci[18].
Facteurs favorisants
La pénétrance assez faible des anomalies génétiques ont fait rechercher des facteurs favorisants autres. Ainsi l'exercice physique (endurance) semble favoriser l'atteinte cardiaque[19].
Il existe également probablement un facteur viral ainsi qu'un facteur auto-immun et un mécanisme inflammatoire semble présent[20].
Diagnostic
Signes fonctionnels
Les premiers signes apparaissent généralement au cours de l'adolescence. Cependant, des signes de dysplasie ventriculaire droite arythmogène ont également été décrits dès le plus jeune âge (chez l'enfant).
Les signes fonctionnels les plus courants sont des palpitations, des syncopes, des signes de défaillance du ventricule droit (œdèmes des membres inférieurs, turgescence jugulaire, reflux hépato-jugulaire).
Une mort subite peut survenir sans aucun signe clinique au préalable et être ainsi malheureusement la première manifestation de la maladie. Elle pourrait ainsi être la cause du quart des morts subites inexpliquées chez l'adulte jeune[21].
Examens complémentaires
Un électrocardiogramme est anormal dans 90 % des cas. L'anomalie la plus fréquente est une inversion de l'onde T dans les dérivations V1 à V3 en l'absence de bloc de branche droit[22]. Cependant, cette anomalie n'est pas spécifique. Un bloc de branche droit peut également être vu. Ceci serait dû à un retard d'activation du ventricule droit, plutôt qu'à une lésion du faisceau droit issu du faisceau de His. Une « onde Epsilon » est trouvée dans 50 % des cas. Elle est décrite comme une « encoche » à la fin des complexes QRS mais elle est difficile à identifier[23]. Elle est le reflet de la conduction intraventriculaire ralentie.
L'enregistrement de l'électrocardiogramme sur 24 heures (holter) peut montrer des troubles du rythme ventriculaire, typiquement d'aspect de retard gauche (c'est-à-dire débutant au niveau du ventricule droit et dépolarisant le ventricule gauche dans un second temps).
L'épreuve d'effort peut démasquer certaines anomalies de manière inconstante. Elle provoque essentiellement des extrasystoles ventriculaires caractéristiques, plus rarement une onde epsilon ou un empâtement de la fin du QRS[24].
Une échographie cardiaque[25] met en évidence une ventricule droit hypokinétique (peu mobile), à la paroi libre très amincie. La dilatation du ventricule droit va entraîner la dilatation de l'orifice de la valve tricuspide, avec une régurgitation (reflux) conséquente de sang au travers la valve tricuspide. Un mouvement paradoxal du septum peut également être présent. Il existe également une dilatation de la voie d'éjection (vers l'artère pulmonaire).
L'IRM cardiaque est devenu l'examen de référence[26], permettant de mesurer la taille et d'estimer la fonction du ventricule droit, et surtout, de retrouver des lésions caractéristiques (infiltration graisseuse et réhaussement tardif à l'injection de gadolinium[27]). Le tissu adipeux prend la forme d'un hypersignal en pondération T1. Cependant, il peut être difficile de différencier la graisse intramyocardique (située à l'intérieur du myocarde) et la graisse épicardique, présente de façon normale autour du cœur, et l'interprétation peut varier selon l'examinateur[28]. Cet examen peut également visualiser l'amincissement extrême et l'akinésie de la paroi libre du ventricule droit. L'injection de gadolinium permet de visualiser la fibrose et améliore substantiellement la précision du diagnostic[29]. Les anomalies cinétiques et électriques de la dysplasie arythmogène du ventricule droit précèdent les anomalies IRM : l'évaluation de la morphologie cardiaque à l'IRM ne semble utile qu'en cas d'anomalie électrique sous-jacente[30]. Toutefois, l'IRM a un intérêt pronostic, les formes biventriculaires ou gauches prédominantes étant plus péjoratives[31].
L'angiographie du ventricule droit, réalisé au cours d'un cathétérisme cardiaque avec injection d'un produit de contraste dans le ventricule droit, peut montrer des anomalies caractéristiques de cette cavité : déformation en « pile d'assiettes ». Elle n'est plus guère pratiquée depuis l’avènement de l'imagerie non invasive.
Le scanner peut retrouver les mêmes types d'anomalies.
Une biopsie transveineuse du ventricule droit peut être spécifique, mais peu sensible. De faux-positifs existent, par exemple lors d'un alcoolisme chronique ou au cours de la dystrophie musculaire de Duchenne. De faux-négatifs sont également possibles, car la maladie progresse typiquement de l'épicarde vers l'endocarde d'une part et d'autre part, de par la nature segmentaire de la maladie (qui n'atteint que certaines zones du myocarde). De plus, de par la finesse de la paroi libre du ventricule droit, beaucoup de prélèvements sont réalisés au niveau du septum inter-ventriculaire, qui lui, n'est habituellement pas atteint par la maladie. Une biopsie compatible avec le diagnostic devrait comporter plus de 3 % de tissu adipeux, plus de 40 % de tissu fibreux et moins de 45 % de myocytes. Le dosage d'une protéine, la plakoglobine, intervenant dans la structure du cytosquelette, dans l'échantillon biopsiée, pourrait être un indice important en faveur de la maladie s'il est abaissé[32].
La place des tests génétiques n'est pas clairement établie. Ils sont utilisés essentiellement dans un but de recherche[8].
L'exploration électrophysiologique avec tentative de déclenchement d'une tachycardie ventriculaire serait d'un intérêt limité pour stratifier le risque de mort subite[8]. Elle reste, toutefois, un argument de bon pronostic si elle se révèle être négative[33].
Critères diagnostiques
Il n'existe pas de signe pathognomonique (qui est caractéristique d'une seule maladie donnée et qui permet d'en établir le diagnostic avec certitude). Le diagnostic est établi, depuis 1994, sur la combinaison de critères majeurs et mineurs. Le diagnostic requiert deux critères majeurs, ou un critère majeur et deux critères mineurs, ou quatre critères mineurs[34].
Critères majeurs
- Dysfonction du ventricule droit :
- myocardiopathie hypokinétique dilatée : dilatation sévère et réduction de la fraction d'éjection (différence entre le volume ventriculaire télédiastolique, en fin de diastole, et le volume télésystolique, en fin de systole, rapporté au volume ventriculaire télédiastolique) du ventricule droit avec ou sans retentissement sur le ventricule gauche ;
- anévrismes localisés du ventricule droit ; classiquement majorés en systole.
- dilatation segmentaire majeure du ventricule droit ;
- Caractéristiques tissulaires : remplacement du myocarde par le tissu fibro-adipeux sur la biopsie myocardique,
- Anomalies de la conduction sur l'électrocardiogramme :
- onde Epsilon en V1-V3 ;
- onde T négative en V1-V3 chez les sujets de plus de 14 ans en l'absence de bloc de branche droit complet ;
- Tachycardie ventriculaire avec aspect de bloc de branche gauche et d'axe supérieur (négatif ou indéterminé en II, III, aVF et positif en aVL)
- Histoire familiale : maladie familiale confirmée à l'autopsie ou lors de la chirurgie.
Critères mineurs
- Dysfonction du ventricule droit :
- dilatation globale modérée du ventricule droit et/ou diminution de la fonction d'éjection avec un ventricule gauche normal ;
- dilatation segmentaire modérée du ventricule droit ;
- hypokinésie localisée du ventricule droit.
- Caractéristiques tissulaires.
- Anomalies de conduction à l'électrocardiogramme :
- extrasystoles fréquentes (plus de 500 par 24h sur le Holter)
- Histoire familiale :
- histoire familiale de mort subite survenue à moins de 35 ans ;
- histoire familiale de dysplasie ventriculaire droite arythmogène.
En cas d'atteinte d'un membre de la famille
Les critères ci-dessus ont été jugés peu sensibles pour la détection de cas précoces, avant toute manifestation clinique, chez les parents au premier degré d'un cas avéré. Ces critères ont donc été modifiés en 2002[35]. La présence d'une onde T négative dans les dérivations antérieures sur l'électrocardiogramme, ou de potentiels tardifs sur l'ECG à haute amplification, ou d'une tachycardie ventriculaire avec aspect de retard gauche, ou la présence de plus de 200 extrasystoles ventriculaires par 24 heures sur le holter, ou l'objectivation d'anomalies du ventricule droit à l'imagerie, suffit à établir une forte présomption de diagnostic.
Diagnostic différentiel
Le syndrome de Brugada, la dysplasie ventriculaire droite arythmogène, la tachycardie ventriculaire polymorphe catécholergique et la naissance de la coronaire droite naissant du sinus coronaire gauche doivent être recherchés systématiquement en cas d'antécédent familial de mort brutale chez un sujet jeune surtout lors d'un effort.
La dysplasie ventriculaire droite arythmogène a longtemps été confondue avec l'anomalie d'Uhl mais ces deux entités sont en réalité bien distinctes.
Évolution et complications
Les complications sont principalement les troubles du rythme cardiaque (tachycardie et fibrillation ventriculaire, fibrillation auriculaire), les risques thromboemboliques, la défaillance cardiaque.
La complication la plus redoutable est la mort subite. La mortalité reste toutefois faible dans une population non sélectionnée, de l'ordre de 1 % par an[36]. Les critères de haut risque de survenue d'une mort subite sont le jeune âge, le sexe mâle[37], une activité sportive en compétition, une histoire familiale de mort subite ou de dysplasie ventriculaire droite arythmogène, une maladie extensive du ventricule droit avec une fraction d'éjection du ventricule droit diminuée, une atteinte du ventricule gauche, la survenue de syncopes, des épisodes d'arythmies ventriculaires[38], la survenue d'une fibrillation atriale[37], la présence d'une mutation sur le gène TMEM43[39]. Une activité sportive semble majorer également ce risque[40]. Un modèle permet de calculer le risque de survenue d'une mort subite[41] et aide dans la discussion de l'indication de la pose d'un défibrillateur automatique implantable[42]. Ce modèle est cependant d'intérêt plus limité en absence de gène responsable identifié[43].
Prise en charge
La prise en charge a fait l'objet de la publication de recommandations de 2015[36]. Le but du traitement de cette maladie est de diminuer l'incidence de mort subite. Il peut être de type médicamenteux, chirurgical. La pose d'un défibrillateur automatique implantable est souvent discutée.
Dans tous les cas, une activité sportive est déconseillée[8], surtout si elle est de type compétition ou réclamant une endurance[36].
Le traitement médicamenteux inclut la prise en charge des troubles du rythme et la prévention de la formation de thrombi (caillots). Le sotalol, bêta-bloquant et antiarythmique de classe III, est le médicament le plus utilisé dans cette maladie mais son efficacité est discutable[44]. D'autres antiarythmiques sont également utilisés, tel l'amiodarone et les bêta-bloquants « traditionnels »[36] comme le metoprolol.
Les sujets présentant une fraction d'éjection du ventricule droit diminuée avec des zones dyskinétiques doivent être mis sous traitement anticoagulant au long cours.
L'implantation d'un défibrillateur automatique est un des éléments de prévention de la survenue d'une mort subite. Ses indications sont : un arrêt cardiaque dû à une tachycardie ventriculaire ou une fibrillation ventriculaire, une atteinte sévère du ventricule droit avec mauvaise tolérance d'une tachycardie ventriculaire, une mort subite chez un parent proche. Dans ces cas, l'implantation de ce dispositif permet de diminuer significativement la mortalité[45].
L'isolation ou la destruction des zones malades par ablation par radiofréquence permet la diminution des complications rythmiques, sans toutefois les annuler[46]. Cette technique est donc essentiellement proposé en cas de chocs répétés par le défibrillateur malgré un traitement médical jugé optimal[36].
La transplantation cardiaque est une option rarement proposée dans cette maladie.
Notes et références
- Rampazzo A, Nava A, Danieli GA, Buja G, Daliento L, Fasoli G, Scognamiglio R, Corrado D, Thiene G (1994) The gene for arrhythmogenic right ventricular cardiomyopathy maps to chromosome 14q23-q24. Hum Mol Genet 3:959-62
- (la) Lancisi GM « De Motu Cordis et Aneurysmatibus Opus Posthumum In Duas Partes Divisum » Naples, 1736
- Frank R, Fontaine G, Vedel J, Mialet G, Sol C, Guiraudon G, Grosgogeat Y. « Électrocardiologie de quatre cas de dysplasie ventriculaire droite arythmogène » Arch Mal Cœur. 1978;71:963
- Vedel J, Frank R, Fontaine G, Dobrinski G, Guiraudon G, Brocheriou C, Grosgogeat Y. « Tachycardies ventriculaires récidivantes et ventricule droit papyracé de l'adulte (à propos de deux observations anatomo-cliniques) » Arch Mal Cœur. 1978:71;973
- Fontaine G, Frank R, Guiraudon G et al. « Signification des troubles de conduction intraventriculaire observés dans la dysplasie ventriculaire droite arhythmogène » Arc Mal Cœur. 1984;77:872-879
- Sandy N. Shah, Krishna Kishore Umapathi et Tony I. Oliver, « Arrhythmogenic Right Ventricular Cardiomyopathy », dans StatPearls, StatPearls Publishing, (PMID 29262224, lire en ligne)
- Mohamed Elmaghawry, Mohammed Alhashemi, Alessandro Zorzi et Magdi H Yacoub, « A global perspective of arrhythmogenic right ventricular cardiomyopathy », Global Cardiology Science & Practice, vol. 2012, no 2, , p. 81–92 (ISSN 2305-7823, PMID 24688993, PMCID 3963715, DOI 10.5339/gcsp.2012.26, lire en ligne, consulté le )
- (en) Basso C, Corrado D, Marcus FI, Nava A, Thiene G, Arrhythmogenic right ventricular cardiomyopathy, Lancet, 2009;373:1289-1300
- (en) Mallat Z, Tedjui A, Fontaliran F, Frank R, Durigon M, Fontaine G. Evidence of apoptosis in arrhythmogenic right ventricular dysplasia, N Engl J Med, 1996;335:1190-1196
- (en) Saffitz JE, Dependence of electrical coupling on mechanical coupling in cardiac myocytes: insights gained from cardiomyopathies caused by defects in cell-cell connections, Ann N Y Acad Sci. 2005;1047:336-344
- Caforio ALP, Re F, Avella A et al. Evidence from family studies for autoimmunity in arrhythmogenic right ventricular cardiomyopathy: Associations of circulating anti-heart and anti-intercalated disk autoantibodies with disease severity and family history, Circulation, 2020;141:1238-1248
- Pérez-Hernández M, van Opbergen CJ, Bagwan N et al. Loss of nuclear envelope integrity and increased oxidant production cause DNA damage in adult hearts deficient in PKP2: A molecular substrate of ARVC, Circulation, 2022;146:851–867
- (en) Beffagna G, Occhi G, Nava A et al. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1, Cardiovasc Res. 2005;65:366–373
- (en) Tiso N, Stephan DA, Nava A et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2), Hum Mol Genet. 2001;10:189–194
- (en) Merner ND, Hodgkinson KA, Haywood AF et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene, Am J Hum Genet. 2008;82:809–821
- (en) Taylor M, Graw S, Sinagra G et al. Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy–overlap Syndromes, Circulation 2011;124:876-885
- (en) Protonotarios N, Tsatsopoulou A, Patsourakos P et al. Cardiac abnormalities in familial palmoplantar keratosis, Br Heart J. 1986;56:321-326
- (en) Kirchhof P, Fabritz L, Zwiener M et al. Age- and training-dependent development of arrhythmogenic right ventricular cardiomyopathy in heterozygous plakoglobin-deficient mice, Circulation 2006; 114:1799-1806
- James CA, Bhonsale A, Tichnell C et al. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers, J Am Coll Cardiol, 2013;62:1290–1297
- Asatryan B, Asimaki An Landstrom AP et al. Inflammation and immune response in arrhythmogenic cardiomyopathy: State-of-the-art review, Circulation, 2021;144:1646–1655
- Thiene G, Nava A, Corrado D, Rossi L, Pennelli N, Right ventricular cardiomyopathy and sudden death in young people, N Engl J Med, 1988;318:129-133
- (en) Jain R, Dalal D, Daly A et al. Electrocardiographic features of arrhythmogenic right ventricular dysplasia, Circulation 2009;120:477-487
- Platonov PG, Calkins H, Hauer RN et al. High interobserver variability in the assessment of epsilon waves: implications for diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia, Heart Rhythm, 2016;13:208–221
- Perrin MJ, Angaran P, Laksman Z et al. Exercise testing in asymptomatic gene carriers exposes a latent electrical substrate of arrhythmogenic right ventricular cardiomyopathy, J Am Coll Cardiol, 2013;62:1772-1779.
- (en) Yoerger DM, Marcus F, Sherrill D et al. Multidisciplinary study of right ventricular dysplasia investigators. Echocardiographic findings in patients meeting task force criteria for arrhythmogenic right ventricular dysplasia: new insights from the multidisciplinary study of right ventricular dysplasia, J Am Coll Cardiol. 2005;45:860-865
- Corrado D, van Tintelen PJ, McKenna WJ et al. Arrhythmogenic right ventricular cardiomyopathy: evaluation of the current diagnostic criteria and differential diagnosis, Eur Heart J, 2020;41:1414–1429
- Aquaro GD, Barison A, Todiere G et al. Usefulness of combined functional assessment by cardiac magnetic resonance and tissue characterization versus task force criteria for diagnosis of arrhythmogenic right ventricular cardiomyopathy, Am J Cardiol, 2016;118:1730–1736
- (en) Bluemke DA, Krupinski EA, Ovitt T et al. MR Imaging of arrhythmogenic right ventricular cardiomyopathy: morphologic findings and interobserver reliability, Cardiology, 2003;99:153-162
- Cardiovascular magnetic resonance in arrhythmogenic right ventricular cardiomyopathy revisited: comparison with task force criteria and genotype, J Am Coll Cardiol. 2006;48:2132-2140
- te Riele ASJM, Bhonsale A, James CA et al. Incremental value of cardiac magnetic resonance imaging in arrhythmic risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy–associated desmosomal mutation carriers, J Am Coll Cardiol, 2013;62:1761–1769
- Aquaro GD, De Luca A, Cappelletto C et al. Prognostic value of magnetic resonance phenotype in patients with arrhythmogenic right ventricular cardiomyopathy, J Am Coll Cardiol, 2020;75:2753-2765
- (en) Asimaki A, Tandri H, Huang H et al. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy, N Eng J Med. 2009;360:1075-1084
- Gasperetti A, Carrick RT, Costa S et al. Programmed ventricular stimulation as an additional primary prevention risk stratification tool in arrhythmogenic right ventricular cardiomyopathy: A multinational study, Circulation, 2022;146:1434–1443
- (en) McKenna WJ, Thiene G, Nava A et al. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology « Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy » Br Heart J. 1994;71:215-218
- (en) Hamid MS, Norman M, Quraishi A et al. Prospective evaluation of relatives for familial arrhythmogenic right ventricular cardiomyopathy/dysplasia reveals a need to broaden diagnostic criteria, J Am Coll Cardiol. 2002;40:1445-1450
- Corrado D, Wichter T, Link MS et al. Treatment of arrhythmogenic right ventricular cardiomyopathy/dysplasia: An international task force consensus statement, Circulation, 2015;132:441-453
- Mazzanti A, Ng K, Faragli A et al. Arrhythmogenic right ventricular cardiomyopathy: clinical course and predictors of arrhythmic risk, J Am Coll Cardiol, 2016;68:2540–2550
- (en) Hulot JS, Jouven X, Empana JP, Frank R, Fontaine G, Natural history and risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy, Circulation 2004;110:1879-1884
- Calkins H, Corrado D, Marcus F, Risk stratification in arrhythmogenic right ventricular cardiomyopathy, Circulation, 2017;136:2068-2082
- (en) Corrado D, Basso C, Rizzoli G, Schiavon M, Thiene G, Does sports activity enhance the risk of sudden death in adolescents and young adults?, J Am Coll Cardiol. 2003;42:1959-1963
- Cadrin-Tourigny J, Bosman LP, Nozza A et al. A new prediction model for ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy, Eur Heart J, 2019;40:1850–1858
- Jordà P, Bosman LP, Gasperetti A et al. Arrhythmic risk prediction in arrhythmogenic right ventricular cardiomyopathy: external validation of the arrhythmogenic right ventricular cardiomyopathy risk calculator, European Heart Journal, 2022;43:3041–3052
- Protonotarios A, Bariani R, Cappelletto C et al. Importance of genotype for risk stratification in arrhythmogenic right ventricular cardiomyopathy using the 2019 ARVC risk calculator, Eur Heart J, 2022;43:3053–3067
- (en) Marcus GM, Glidden DV, Polonsky B et al. Efficacy of antiarrhythmic drugs in arrhythmogenic right ventricular cardiomyopathy: A report from the North American ARVC Registry, J Am Coll Cardiol. 2009;54:609-615
- (en) Corrado D, Leoni L, Link MS et al. Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia, Circulation 2003;108:3084-3091
- Philips B, Madhavan S, James C et al. Outcomes of catheter ablation of ventricular tachycardia in arrhythmogenic right ventricular dysplasia/cardiomyopathy, Circ Arrhythm Electrophysiol, 2012;5:499–505