Structure secondaire d'un acide nucléique
La structure secondaire d'un acide nucléique correspond à la conformation obtenue par les interactions entre les paires de bases au sein d'un seul polymère d'acide nucléique ou bien entre deux de ces polymères. Cette structure peut être représentée comme une liste de bases appariées avec une molécule d'acide nucléique[1]. Les structures secondaires des ADN et ARN biologiques ont tendance à être différentes : l'ADN biologique existe en majorité sous forme de double hélice où toutes les bases sont appariées, alors que l'ARN biologique est simple brin et forme souvent des interactions entre paires de bases assez complexes en raison de sa capacité accrue à former des liaisons hydrogène issues du groupe hydroxyle extérieur au niveau de chaque ribose.
.png.webp)
Dans un contexte non biologique, la structure secondaire constitue une donnée importante à considérer dans la conception de structures d'acides nucléiques dans le domaine des nanotechnologies en ADN et des ordinateurs à ADN, car le motif d'appariement de bases détermine la structure globale des molécules.
Concepts fondamentaux
Appariement des bases


En biologie moléculaire, deux nucléotides situés sur des brins complémentaires d'ADN ou d'ARN et liés par des liaisons hydrogène sont appelés "paire de bases" (souvent abrégé en pb, ou bp pour base pair en anglais). Dans l'appariement canonique de Watson et Crick, l'adénine (A) forme une paire de bases avec la thymine (T), et la guanine (G) forme une paire de bases avec la cytosine (C) dans l'ADN. Dans l'ARN, la thymine est remplacée par l'uracile (U). Des motifs de liaisons hydrogène alternatifs, comme le wobble pairing et l'appariement Hoogsteen, peuvent également avoir lieu — particulièrement dans l'ARN —, ce qui donne naissance à des structures tertiaires fonctionnelles et complexes. L'appariement est important car il constitue le mécanisme par lequel les codons sur l'ARNm sont reconnus par des anticodons des ARNt au cours de la traduction en protéines. Certaines enzymes de liaison à l'ADN ou l'ARN peuvent reconnaître des motifs d'appariement spécifiques qui permettent d'identifier des régions particulières de régulation des gènes. Le phénomène de liaison hydrogène est le mécanisme chimique sous-jacent aux règles d'appariement de base décrites plus haut. Une correspondance géométrique appropriée entre les donneurs et accepteurs de liaisons hydrogène permet seulement aux "bonnes" paires de se former de façon stable. L'ADN ayant un important contenu en GC est plus stable que l'ADN avec un contenu en GC plus faible, mais contrairement à une idée reçue les liaisons hydrogène ne stabilisent pas l'ADN de façon significative ; cette stabilisation est principalement due à des interactions d'empilement[2].
Les bases les plus grandes en taille, à savoir l'adénine et la guanine, sont membres d'une classe de structures chimiques en double anneau qu'on appelle des purines ; les bases les plus petites, à savoir la cytosine et thymine (et l'uracile), sont membres d'une classe de structures chimiques en anneau simple qu'on appelle des pyrimidines. Les purines sont complémentaires seulement avec les pyrimidines : les appariements pyrimidine-purine sont énergétiquement défavorables parce que les molécules sont trop distantes pour pouvoir établir une liaison hydrogène ; les appariements purine-purine sont énergétiquement défavorables parce que les molécules sont trop proches, ce qui entraîne une répulsion de chevauchement. Les seuls autres appariements possibles sont GT et AC ; ces appariements sont mal assortis parce que le motif des donneurs et accepteurs d'hydrogène ne correspondent pas. La wobble pair GU, avec deux liaisons hydrogène, ne se produit pas très souvent dans l'ARN.
Hybridisation d'acide nucléique
L'hybridisation est le processus de liaison de paires de bases complémentaires pour former une double hélice. La fusion est le processus par lequel les interactions entre les brins de la double hélice sont brisées, ce qui sépare les deux brins de l'acide nucléique. Ces liaisons sont faibles, facilement séparées par une légère augmentation de la température, des enzymes ou une force physique. La fusion a préférentiellement lieu à certains points au niveau de l'acide nucléique[3]. Les séquences riches en T et A sont plus sensibles à la fusion que les régions riches en C et G. Des étapes de base particulières sont aussi sensibles à la fusion de l'ADN, en particulier les étapes T-A et T-G[4]. Ces caractéristiques mécaniques sont mises en évidence par l'utilisation de séquences comme les boîtes TATA en début de nombreux gènes afin d'aider l'ARN polymérase à "fondre" l'ADN pour initier la transcription.
La séparation des brins par chauffement léger, comme on le fait en PCR, est simple à condition que les molécules aient moins de 10 000 paires de bases (10 kilo(paires de) bases, ou 10 k(p)b). L'entrelacement des brins d'ADN rend les longs segments difficiles à séparer. La cellule contourne ce problème en permettant à ses enzymes de fusion de l'ADN (hélicases) à travailler en parallèle avec les topoisomérases, qui peuvent chimiquement cliver l'ossature phosphate de l'un des brins de telle façon qu'il puisse pivoter autour de l'autre. Les hélicases déroulent les brins pour faciliter l'avancée d'enzymes lisant les séquences comme l'ADN polymérase.
Motifs des structures secondaires
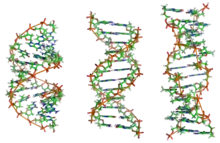
La structure secondaire d'un acide nucléique est généralement divisée en deux hélices (paires de bases contiguës), et diverses sortes de boucles (nucléotides non appariés entourés d'hélices). Fréquemment, ces éléments, ou une combinaison de ces éléments, peuvent ensuite être classifié, par exemple les tétraboucles, les pseudonœuds et les tiges-boucles.
Double hélice
La double hélice est une importante structure tertiaire chez les molécules d'acides nucléiques, intimement connectée avec la structure secondaire de la molécule. Une double hélice est formée par des régions composées de plusieurs paires de bases consécutives.
La double hélice d'acide nucléique est un polymère en spirale, généralement dextrogyre, contenant deux brins de nucléotides dont les paires de bases s'apparient entre elles. Un simple tour de l'hélice fait environ 10 nucléotides, et contient un creux majeur et un creux mineur, le majeur étant plus large que le mineur[5]. Compte tenu de cette différence, de nombreuses protéines qui se lient à l'ADN le font au niveau du creux majeur[6]. Plusieurs formes de double hélice sont possibles ; pour l'ADN, les trois formes biologiquement pertinentes sont l'ADN A, l'ADN B et l'ADN Z, alors que les doubles hélices d'ARN ont des structures similaires à la forme A de l'ADN.
Tiges-boucles
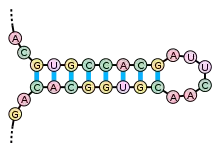
La structure secondaire des molécules d'acides nucléiques peut souvent être décomposée uniquement en tiges et en boucles. La structure en tige-boucle, dans laquelle une hélice contenant des bases appariées se termine en une petite boucle non appariée, est extrêmement commune et constitue un bloc de construction pour des motifs structurels plus grands tels que les structures en feuille de trèfle, qui sont des jonctions de quatre hélices comme celles qu'on trouve chez les ARNt. Les boucles internes (petites séries de bases non appariées dans une hélice contenant plus de bases appariées) et les bosses (régions où un brin d'une hélice a des "extra" bases insérées sans équivalent dans le brin opposé) sont également fréquentes.
Il y a beaucoup d'éléments de structure secondaire d'importance fonctionnelle dans les ARN biologiques ; certains exemples connus sont les tiges-boucles avec terminaisons intrinsèques et la feuille de trèfle de l'ARNt. Il existe encore une petite communauté de chercheurs qui essaie de déterminer la structure secondaire des molécules d'ARN. Les approches incluent à la fois des méthodes expérimentales et informatiques.
Pseudonœuds
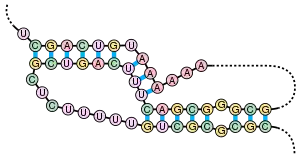
Un pseudonœud est une structure secondaire d'acide nucléique contenant au moins deux structures en tige-boucle dans laquelle la moitié d'une tige est intercalée entre les deux moitiés de l'autre tige. Les pseudonœuds se replient en des conformations tridimensionnelles en forme de nœud mais ne sont pas de vrais nœuds topologiques. L'appariement des bases dans les pseudonœuds n'est pas bien imbriqué, c'est-à-dire qu'il arrive que des paires de bases se "chevauchent" en position de séquence. Cela rend la présence de pseudonœuds dans les séquences d'acides nucléiques impossible à prédire par la méthode standard de programmation dynamique, qui utilise un système de scoring récursif pour identifier des tiges appariées, et en conséquence ne peut pas détecter des paires de bases non imbriquées avec les algorithmes les plus communs. Des sous-classes limitées de pseudonœuds peuvent être prédites en utilisant des programmes dynamiques décrits dans la référence ci-indiquée[8]. De nouvelles techniques de prédiction de structure comme les grammaires stochastiques sans contexte ne prennent également pas en compte les pseudonœuds.
Les pseudonœuds possèdent une variété de structures avec une activité catalytique[9] et plusieurs processus biologiques importants sont basés sur des molécules d'ARN qui forment des pseudonœuds. Par exemple, le composant ARN de la télomérase humaine contient un pseudonœud crucial pour son activité enzymatique[7]. Le ribozyme du virus de l'hépatite D est un exemple connu d'ARN catalytique avec un pseudonœud dans son site actif[10] - [11]. Bien que l'ADN puisse aussi former des pseudonœuds, on ne les retrouve généralement pas dans l'ADN biologique.
Prédiction de la structure secondaire
La plupart des méthodes de prédiction de la structure secondaire des acides nucléiques est basée sur un modèle d'énergie du voisin le plus proche[12] - [13]. Une méthode générale pour calculer la structure secondaire probable d'un acide nucléique est la programmation dynamique, qui est utilisée pour calculer des structures en optimisant l'énergie libre thermodynamique. Les algorithmes de programmation dynamique excluent souvent les pseudonœuds ou d'autres cas dans lesquels les paires de bases ne sont pas totalement imbriquées, car le fait de prendre en compte ces structures devient informatiquement très cher, même pour les petits acides nucléiques. D'autres méthodes, comme les grammaires stochastiques sans contexte, peuvent aussi être utilisée pour prédire la structure secondaires des acides nucléiques.
Pour beaucoup de molécules d'ARN, la structure secondaire est très importante pour assurer correctement la fonction de l'ARN — souvent plus que la séquence en elle-même (structure primaire). Ce fait aide à analyser l'ARN non codant, parfois désigné "gènes d'ARN". Une application de la bioinformatique utilise des structures secondaires d'ARN prédites pour rechercher des formes non codantes mais fonctionnelles d'un ARN au sein d'un génome. Par exemple, les micro-ARN ont des structures en tiges-boucles longues canoniques, entrecoupées de petites boucles internes.
La structure secondaire d'ARN s'applique dans l'épissage chez certaines espèces. Chez les humains et les autres tétrapodes, on a montré que sans la protéine U2AF2, le processus d'épissage est inhibé. Cependant, chez le poisson-zèbre et d'autres téléostéens, le processus d'épissage de l'ARN peut toujours avoir lieu au niveau de certains gènes en l'absence de cette protéine. Cela peut être parce que 10% des gènes du poisson-zèbre ont des paires de bases alternant TG et AC aux sites d'épissage 3' et 5' respectivement sur chaque intron, ce qui altère la structure secondaire de l'ARN. Cela suggère que la structure secondaire de l'ARN peut influencer l'épissage, potentiellement sans l'utilisation de protéines telles que U2AF2 dont on pensait qu'elles étaient requises pour que l'épissage ait lieu[14].
Voir aussi
Notes
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Nucleic acid secondary structure » (voir la liste des auteurs).
Articles connexes
Liens externes
- (en) Base de données DiProDB : permet de collecter et d'analyser des propriétés de dinucléotides telles que leur thermodynamique, leur structure, etc.
Références
- Robert M. Dirks, Milo Lin, Erik Winfree et Niles A. Pierce, « Paradigms for computational nucleic acid design », Nucleic Acids Research, vol. 32, no 4, , p. 1392–1403 (ISSN 1362-4962, PMID 14990744, DOI 10.1093/nar/gkh291, lire en ligne, consulté le )
- Peter Yakovchuk, Ekaterina Protozanova et Maxim D. Frank-Kamenetskii, « Base-stacking and base-pairing contributions into thermal stability of the DNA double helix », Nucleic Acids Research, vol. 34, no 2, , p. 564–574 (ISSN 1362-4962, PMID 16449200, PMCID PMC1360284, DOI 10.1093/nar/gkj454, lire en ligne, consulté le )
- K. J. Breslauer, R. Frank, H. Blöcker et L. A. Marky, « Predicting DNA duplex stability from the base sequence », Proceedings of the National Academy of Sciences of the United States of America, vol. 83, no 11, , p. 3746–3750 (ISSN 0027-8424, PMID 3459152, lire en ligne, consulté le )
- (en) Richard Owczarzy, « DNA melting temperature - How to calculate it? », High-throughput DNA biophysics, (lire en ligne)
- (en) Bruce Alberts et al., The Molecular Biology of the Cell, New York, Garland Science, (ISBN 978-0-8153-4105-5)
- C. O. Pabo et R. T. Sauer, « Protein-DNA recognition », Annual Review of Biochemistry, vol. 53, , p. 293–321 (ISSN 0066-4154, PMID 6236744, DOI 10.1146/annurev.bi.53.070184.001453, lire en ligne, consulté le )
- Jiunn-Liang Chen et Carol W. Greider, « Functional analysis of the pseudoknot structure in human telomerase RNA », Proceedings of the National Academy of Sciences of the United States of America, vol. 102, no 23, , p. 8080–8085; discussion 8077–8079 (ISSN 0027-8424, PMID 15849264, PMCID PMC1149427, DOI 10.1073/pnas.0502259102, lire en ligne, consulté le )
- E. Rivas et S. R. Eddy, « A dynamic programming algorithm for RNA structure prediction including pseudoknots », Journal of Molecular Biology, vol. 285, no 5, , p. 2053–2068 (ISSN 0022-2836, PMID 9925784, DOI 10.1006/jmbi.1998.2436, lire en ligne, consulté le )
- David W. Staple et Samuel E. Butcher, « Pseudoknots: RNA structures with diverse functions », PLoS biology, vol. 3, no 6, , e213 (ISSN 1545-7885, PMID 15941360, PMCID PMC1149493, DOI 10.1371/journal.pbio.0030213, lire en ligne, consulté le )
- A. R. Ferré-D'Amaré, K. Zhou et J. A. Doudna, « Crystal structure of a hepatitis delta virus ribozyme », Nature, vol. 395, no 6702, , p. 567–574 (ISSN 0028-0836, PMID 9783582, DOI 10.1038/26912, lire en ligne, consulté le )
- Michael M. C. Lai, « The Molecular Biology of Hepatitis Delta Virus », Annual Review of Biochemistry, vol. 64, no 1, , p. 259–286 (ISSN 0066-4154, DOI 10.1146/annurev.bi.64.070195.001355, lire en ligne, consulté le )
- T. Xia, J. SantaLucia, M. E. Burkard et R. Kierzek, « Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson-Crick base pairs », Biochemistry, vol. 37, no 42, , p. 14719–14735 (ISSN 0006-2960, PMID 9778347, DOI 10.1021/bi9809425, lire en ligne, consulté le )
- David H. Mathews, Matthew D. Disney, Jessica L. Childs et Susan J. Schroeder, « Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure », Proceedings of the National Academy of Sciences of the United States of America, vol. 101, no 19, , p. 7287–7292 (ISSN 0027-8424, PMID 15123812, DOI 10.1073/pnas.0401799101, lire en ligne, consulté le )
- Chien-Ling Lin, Allison J. Taggart, Kian Huat Lim et Kamil J. Cygan, « RNA structure replaces the need for U2AF2 in splicing », Genome Research, vol. 26, no 1, , p. 12–23 (ISSN 1549-5469, PMID 26566657, PMCID PMC4691745, DOI 10.1101/gr.181008.114, lire en ligne, consulté le )