Réaction de Johnson-Corey-Chaykovsky
La réaction de Johnson–Corey–Chaykovsky (parfois appelée réaction de Corey–Chaykovsky) est une réaction utilisée en chimie organique pour la synthèse d'époxydes, d'aziridines et de cyclopropanes. Elle a été découverte en 1961 par A. William Johnson et développée de manière significative par Elias James Corey et Michael Chaykovsky. La réaction implique l'ajout d'un ylure de soufre à une cétone, un aldéhyde, une imine ou une énone pour produire le cycle à trois membres correspondant. La réaction est diastéréosélective, favorisant la substitution trans dans le produit quelle que soit la stéréochimie initiale. La synthèse d'époxydes via cette méthode constitue une alternative rétrosynthétique importante aux réactions d'époxydation traditionnelles des oléfines.
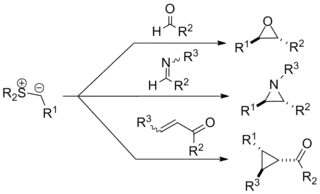
La réaction est le plus souvent employée pour l'époxydation par transfert de méthylène, et à cette fin a été utilisée dans plusieurs synthèses totales notables (voir Synthèse d'époxydes). L'historique, le mécanisme, l'étendue de la réaction et les variantes énantiosélectives de la réaction sont également détaillés ci-dessous. Plusieurs revues de littérature ont été publiées[1] - [2] - [3] - [4] - [5] - [6].
Histoire
La publication originale de Johnson concernait la réaction du fluorénylure de 9-diméthylsulfonium avec des dérivés de benzaldéhyde substitués. La tentative de réaction de type Wittig a échoué et un oxyde de benzalfluorène a été obtenu à la place, notant que, contrairement aux ylures de phosphore et d'arsenic, l'ylure de soufre ne forme pas de benzalfluorènes lorsque mis en réaction avec des benzaldéhydes[7].
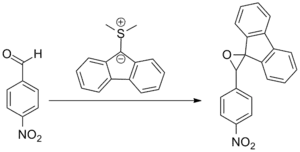
Les réactifs de Corey-Chaykovsky (soit le (CH3)2SOCH2 et le (CH3)2SCH2) par Corey et Chaykovsky, en tant que réactifs efficaces de transfert de méthylène, ont permis de populariser cette réaction[8].
Mécanisme
Le mécanisme réactionnel de la réaction de Johnson-Corey-Chaykovsky consiste en une addition nucléophile de l'ylure au groupe carbonyle ou imine. Une charge négative est transférée à l'hétéroatome et le cation sulfonium expulsé lors de la formation du cycle, puisqu'il est un bon groupe partant. Dans la réaction de Wittig associée, la formation de la double liaison phosphore-oxygène est favorisée, ce qui empêche la formation d'oxirane et mène à l'oléfination via un intermédiaire cyclique à quatre membres[4].

La diastéréosélectivité trans observée résulte de la réversibilité de l'addition initiale, ce qui favorise la formation de la bétaïne anti par rapport à la bétaïne syn. L'ajout initial de l'ylure donne une bétaïne avec des charges adjacentes; les calculs de la théorie de la fonctionnelle de la densité ont montré que l'étape limitant la vitesse est la rotation de la liaison centrale dans le conformère nécessaire pour l'éjection du sulfonium lors de la cyclisation[1].
_V2.png.webp)
Le degré de réversibilité dans l'étape initiale (et donc la diastéréosélectivité) dépend de quatre facteurs, une plus grande réversibilité correspondant à une plus grande sélectivité[1] :
- La stabilité du substrat qui peut être suffisamment élevée pour que l'équilibre favorise les produit de départ par rapport à la bétaïne ;
- La stabilité de l'ylure qui peut, de manière similaire, favoriser les produits de départ ;
- L'encombrement stérique dans la bétaïne qui, avec un encombrement plus important, conduit à une plus grande réversibilité en défavorisant la formation de l'intermédiaire et en ralentissant la vitesse de rotation limitante de la liaison centrale ;
- La solvatation des charges dans la bétaïne par des contre-ions tels que le lithium avec une plus grande solvatation permettant une rotation plus facile dans l'intermédiaire de bétaïne, réduisant le degré de réversibilité.
Étendue de la réaction
L'application de la réaction de Johnson–Corey–Chaykovsky en synthèse organique est diverse. La nature des ylures de soufre et des électrophiles qu'il est possible d'utiliser pour cette réaction s'est élargie. Elle a d'ailleurs été utilisée dans un certain nombre de synthèses totales, comme détaillé ci-dessous, et est généralement reconnu comme un puissant outil de transformation dans le répertoire organique.
Types d'ylures
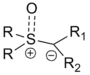
Les ylures de soufre peuvent être préparés avec une grande variété de groupes fonctionnels, et ce autant sur le carbanion que sur le soufre. Le patron de substitution peut influencer la facilité de préparation des réactifs (généralement à partir de l'halogénure de sulfonium, par exemple l'iodure de triméthylsulfonium) et la vitesse de réaction globale de diverses manières. Le format général du réactif est indiqué à droite[1].
La préparation de l'ylure à partir d'un sulfoxonium est plus facile que la préparation à partir d'un sulfonium et permet l'utilisation de bases plus faibles. Cela peut s'expliquer par la présence d'une liaison double entre l'atome de soufre et l'oxygèse alors que le sulfonium n'en contient pas. De plus, les sous-produits de dialkylsulfoxyde des réactifs sulfoxonium sont largement préférés aux sous-produits de dialkylsulfure des réactifs sulfonium qui sont significativement plus toxiques, volatils et odorants[1].
La grande majorité des réactifs sont monosubstitués au niveau du carbanion de l'ylure (soit R1 ou R2 est un hydrogène). Les réactifs disubstitués sont beaucoup plus rares mais ont été décrits[1] :
- Si le carbone de l'ylure est substitué par un groupe électroattracteur (GEA), le réactif est appelé ylure stabilisé. Ceux-ci, comme les réactifs sulfoxonium, réagissent beaucoup plus lentement et sont généralement plus faciles à préparer. Ils sont cependant limités dans leur utilisation car la réaction peut devenir excessivement lente : les exemples impliquant des amides sont répandus, mais il y en a beaucoup moins impliquant des esters et pratiquement aucun exemple impliquant d'autres EGA. Pour ces derniers, la réaction de Darzens associée est généralement plus appropriée ;
- Si le carbone de l'ylure est substitué par un groupe aryle ou allyle, le réactif est appelé ylure semi-stabilisé. Ceux-ci ont été largement développés, et seulement les réactifs méthylène classiques (R1 = R2 = H) sont plus utilisés. La présence de groupes fonctionnels sur les réactifs aryle peut fortement influencer la sélectivité de la réaction selon les critères ci-dessus ;
- Si le carbone de l'ylure est substitué par un groupe alkyle, le réactif est appelé ylure non stabilisé. La taille des groupes alkyles sont les facteurs majeurs de sélectivité avec ces réactifs.
Les groupes R sur le soufre, bien que généralement des méthyles, ont été utilisés pour synthétiser des réactifs qui peuvent effectuer des variantes énantiosélectives de la réaction (voir les variations ci-dessous). La taille des groupes peut également influencer la diastéréosélectivité dans les substrats alicycliques[1].
Synthèse d'époxydes
Les réactions des ylures de soufre avec des cétones et des aldéhydes pour former des époxydes sont de loin l'application la plus courante de la réaction de Johnson–Corey–Chaykovsky. Des exemples impliquant des substrats complexes et des ylures « exotiques » ont été rapportés, comme indiqué ci-dessous[9] - [10].
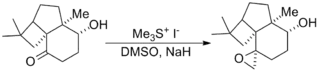
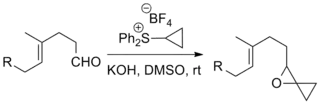
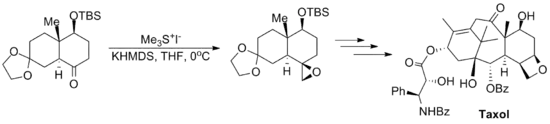
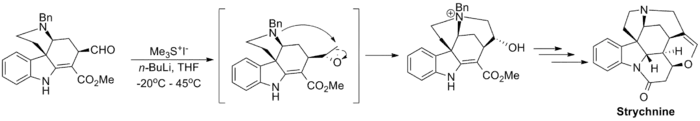
La réaction a été utilisée dans un certain nombre de synthèses totales notables, y compris la synthèse totale du taxol (un médicament chimiothérapeutique) reportée par Danishefsky[11], et la synthèse totale de la strychnine (un pesticide) du groupe de Kuehne[12].
Synthèse des aziridines
La synthèse d'aziridines à partir d'imines est une autre application importante de la réaction de Johnson–Corey–Chaykovsky et offre une alternative au transfert d'amines à partir d'oxaziridines. Bien que moins appliquée, la réaction a une étendue de substrat et une tolérance de groupes fonctionnels similaires à l'équivalent carbonyle. Les exemples ci-dessous sont représentatifs ; dans ce dernier, une aziridine est formée in situ et s'ouvre par attaque nucléophile pour former l'amine correspondante[3] - [9].
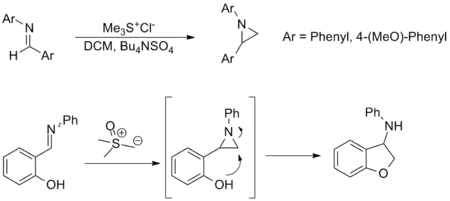
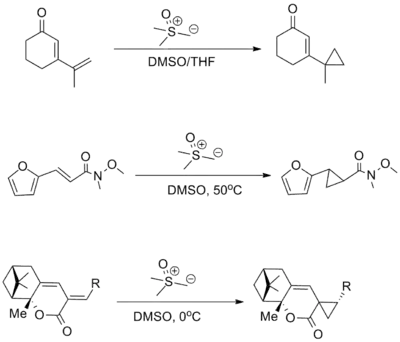
Synthèse de cyclopropanes
Pour l'addition d'ylures de soufre aux énones, une sélectivité 1,4 plus importante est généralement obtenue avec des réactifs sulfoxonium qu'avec des réactifs sulfonium. De nombreux GEA se sont révélés compatibles avec la réaction, comme les cétones, les amides (le deuxième exemple ci-dessous implique un amide de Weinreb) et les esters. Avec d'autres systèmes conjugués, l'addition en 1,6 a tendance à prédominer sur l'addition en 1,4, comme illustré dans le premier exemple ci-dessous[3] - [9].
Autres réactions
En plus des réactions originalement rapportées par Johnson, Corey et Chaykovsky, les ylures de soufre ont été utilisés pour un certain nombre de réactions d'homologation qui sont souvent regroupées sous le même nom.
- Avec les époxydes et les aziridines, la réaction sert à agrandir le cycle pour produire l'oxétane ou l'azétidine correspondant. Toutefois, ce ne sont pas des réactions secondaires observées lors de la synthèse de ces motifs puisque les temps de réaction sont beaucoup plus longs[9].
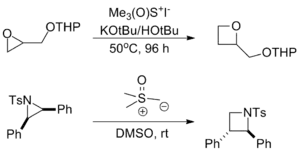
- Plusieurs cycloadditions dans lesquelles l'ylure de soufre agit à titre d'un équivalent de carbénoïde nucléophile ont été reportées[9].
![[4+1] cycloaddition with Corey–Chaykovsky reagent](https://img.franco.wiki/i/CCR41.png.webp)
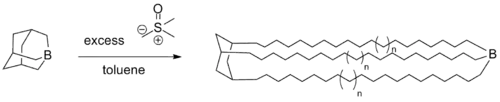
- Des polymérisations vivantes utilisant des trialkylboranes comme catalyseur et du (diméthyloxosulfaniumyl)méthanide comme monomère ont été rapportées pour la synthèse de divers polymères complexes[13].
Variations énantiosélectives
Le développement d'une variante énantiosélective (c'est-à-dire produisant un excès énantiomérique, qui est étiqueté « ee ») de cette réaction demeure un domaine d'actualité de la recherche académique. L'utilisation de sulfures chiraux de manière stœchiométrique s'est avérée plus efficace que l'utilisation de catalyseurs chiraux, mais l'étendue des substrats est encore limitée dans tous les cas. Les organosulfures typiquement utilisés sont généralement abordables et les réactions racémiques peuvent être réalisées avec des quantités équimolaires d'ylure et un catalyseur chiral sans augmenter les coûts de manière significative. Les sulfures chiraux, en revanche, sont plus coûteux à préparer, ce qui stimule l'avancement des méthodes énantiosélectives catalytiques[2].
Réactifs stœchiométriques
Les réactifs les plus efficaces utilisés de manière stœchiométrique sont présentés ci-dessous. Le premier est un oxathiane bicyclique qui a été utilisé dans la synthèse du composé β-adrénergique dichloroisoprotérénol (DCI), mais est limité par la disponibilité d'un seul énantiomère du réactif. La synthèse du diastéréoisomère axial est rationalisée via l'effet 1,3-anomérique qui réduit la nucléophilie de la paire solitaire équatoriale. La conformation de l'ylure est limitée par la tension transannulaire et l'approche de l'aldéhyde est limitée à une face de l'ylure par des interactions stériques avec les substituants méthyle[2] - [5].
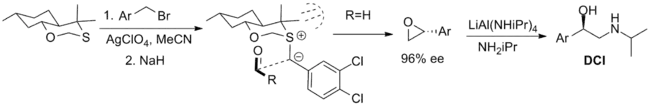
L'autre réactif majeur est un réactif dérivé du camphre développé par Varinder Aggarwal de l'université de Bristol. Les deux énantiomères sont facilement synthétisés, bien que les rendements soient inférieurs à ceux du réactif oxathiane. La conformation de l'ylure est déterminée par l'interaction avec les hydrogènes de la tête de pont et l'approche de l'aldéhyde est bloquée par la fraction camphre. La réaction utilise une base phosphazène pour favoriser la formation de l'ylure[2] - [5].
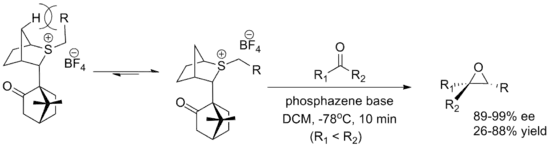
Réactifs catalytiques
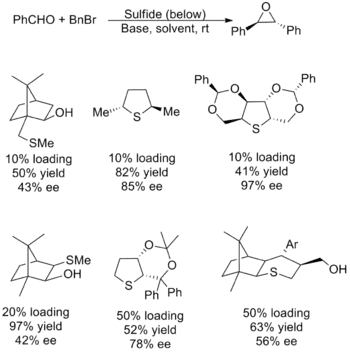
Les réactifs catalytiques ont eu moins de succès, puisque la plupart des réactions énantiosélectives souffrent d'un faible rendement, d'une mauvaise énantiosélectivité, ou des deux. Il existe également des problèmes avec la diversité des substrats, la plupart étant limité au transfert d'un méthylène sur des aldéhydes aliphatiques. Le problème provient du besoin d'un sulfure nucléophile qui génère efficacement l'ylure qui peut également agir comme un bon groupe partant pour former l'époxyde. Étant donné que ces deux facteurs sont en contradiction, il est difficile d'avoir un réactif ayant des propriétés optimales. Plusieurs des catalyseurs les plus performants ainsi que les rendements et l'excès énantiomérique pour leur utilisation dans la synthèse de l'oxyde de (E)-stilbène sont présentés ci-dessous[2] - [5].
Animation du mécanisme

Articles connexes
Références
- (en) Varinder K. Aggarwal et Jeffery Richardson, « The complexity of catalysis: origins of enantio- and diastereocontrol in sulfur ylide mediated epoxidation reactions », Chemical Communications, no 21, , p. 2644 (ISSN 1359-7345 et 1364-548X, DOI 10.1039/b304625g, lire en ligne, consulté le ).
- (en) Varinder K. Aggarwal et Caroline L. Winn, « Catalytic, Asymmetric Sulfur Ylide-Mediated Epoxidation of Carbonyl Compounds: Scope, Selectivity, and Applications in Synthesis », Accounts of Chemical Research, vol. 37, no 8, , p. 611–620 (ISSN 0001-4842 et 1520-4898, DOI 10.1021/ar030045f, lire en ligne, consulté le ).
- (en) Yu.G. Gololobov, A.N. Nesmeyanov, V.P. lysenko et I.E. Boldeskul, « Twenty-five years of dimethylsulfoxonium ethylide (corey's reagent) », Tetrahedron, vol. 43, no 12, , p. 2609–2651 (DOI 10.1016/S0040-4020(01)86869-1, lire en ligne, consulté le ).
- (en) An-Hu Li, Li-Xin Dai et Varinder K. Aggarwal, « Asymmetric Ylide Reactions: Epoxidation, Cyclopropanation, Aziridination, Olefination, and Rearrangement », Chemical Reviews, vol. 97, no 6, , p. 2341–2372 (ISSN 0009-2665 et 1520-6890, DOI 10.1021/cr960411r, lire en ligne, consulté le ).
- (en) Varinder K. Aggarwal, J. Gair Ford, Sílvia Fonquerna et Harry Adams, « Catalytic Asymmetric Epoxidation of Aldehydes. Optimization, Mechanism, and Discovery of Stereoelectronic Control Involving a Combination of Anomeric and Cieplak Effects in Sulfur Ylide Epoxidations with Chiral 1,3-Oxathianes », Journal of the American Chemical Society, vol. 120, no 33, , p. 8328–8339 (ISSN 0002-7863 et 1520-5126, DOI 10.1021/ja9812150, lire en ligne, consulté le ).
- (en) Eoghan M. McGarrigle, Eddie L. Myers, Ona Illa et Michael A. Shaw, « Chalcogenides as Organocatalysts », Chemical Reviews, vol. 107, no 12, , p. 5841–5883 (ISSN 0009-2665 et 1520-6890, DOI 10.1021/cr068402y, lire en ligne, consulté le ).
- (en) A. William Johnson et Robert B. LaCount, « The Chemistry of Ylids. VI. Dimethylsulfonium Fluorenylide—A Synthesis of Epoxides 1 », Journal of the American Chemical Society, vol. 83, no 2, , p. 417–423 (ISSN 0002-7863 et 1520-5126, DOI 10.1021/ja01463a040, lire en ligne, consulté le ).
- (en) E. J. Corey et Michael Chaykovsky, « Dimethyloxosulfonium Methylide ((CH3)2SOCH2) and Dimethylsulfonium Methylide ((CH3)2SCH2). Formation and Application to Organic Synthesis », Journal of the American Chemical Society, vol. 87, no 6, , p. 1353–1364 (ISSN 0002-7863 et 1520-5126, DOI 10.1021/ja01084a034, lire en ligne, consulté le ).
- Jie Jack Li, Name reactions in heterocyclic chemistry, Wiley-Interscience, (ISBN 0-471-70414-8, 978-0-471-70414-0 et 0-471-30215-5, OCLC 57436653, lire en ligne).
- Michael G. Ellerd et Frank G. Favaloro, Name reactions and reagents in organic synthesis, Wiley, (ISBN 978-1-60119-634-7, 1-60119-634-2 et 0-471-22854-0, OCLC 299593042, lire en ligne).
- (en) Samuel J. Danishefsky, John J. Masters, Wendy B. Young et J. T. Link, « Total Synthesis of Baccatin III and Taxol », Journal of the American Chemical Society, vol. 118, no 12, , p. 2843–2859 (ISSN 0002-7863 et 1520-5126, DOI 10.1021/ja952692a, lire en ligne, consulté le ).
- (en) Martin E. Kuehne et Feng Xu, « Total synthesis of strychnan and aspidospermatan alkaloids. 3. The total synthesis of (.+-.)-strychnine », The Journal of Organic Chemistry, vol. 58, no 26, , p. 7490–7497 (ISSN 0022-3263 et 1520-6904, DOI 10.1021/jo00078a030, lire en ligne, consulté le ).
- (en) Jun Luo et Kenneth J. Shea, « Polyhomologation. A Living C1 Polymerization », Accounts of Chemical Research, vol. 43, no 11, , p. 1420–1433 (ISSN 0001-4842 et 1520-4898, DOI 10.1021/ar100062a, lire en ligne, consulté le ).