Pemphigoïde bulleuse
La pemphigoïde bulleuse (PB) est une maladie rare. Il s'agit d'une des affections dermatologiques, dites « dermatoses bulleuses », qui sont des affections subépidermales non-contagieuses caractérisées par des lésions de la peau, en grandes plaques rouges (plaques érythémateuses) garnies de « bulles » (dans ce cas : cloques de couleur jaunâtre épaisses).
| Médicament | Sulfapyridine (en), prednisone, méthotrexate, prednisolone, mycophénolate mofétil (d) et rituximab |
|---|---|
| Spécialité | Dermatologie |
| CIM-10 | L12.0 |
|---|---|
| CIM-9 | 694.5 |
| OMIM | 178500 |
| MedlinePlus | 000883 |
| eMedicine | 1062391 |
| MeSH | D010391 |
| Patient UK | Bullous-pemphigoid-pro |
![]() Mise en garde médicale
Mise en garde médicale
La pemphigoïde bulleuse est d'origine auto-immune et est acquise : elle est due à l'effet des anticorps sur les éléments constitutifs de la peau, comme les desmosomes et hémidesmosomes, protéines de cohésion entre cellules et matrice extracellulaire. Rarement, des muqueuses sont touchées (intérieur de la bouche dans 10 à 20 % des cas).
Cette maladie peut être grave et invalidante. Elle implique un traitement de longue durée.
Épidémiologie
C'est la moins rare parmi les maladies bulleuses auto-immunes.
Sa prévalence (nombre de personnes atteintes dans une population donnée) est d'environ 1 personne sur 40 000 en France (1 personne sur 3 000 à partir de 70 ans)[1].
L'incidence (nouveaux cas par an) est estimée en France à environ 22 cas par millions d'habitants et par an, avec une multiplication par trois des chiffres des années 1990[2]. Chez les patients de plus de 70 ans, elle est de 162 cas par millions d'individus et par an[2]. Les données sont comparables dans le reste de l'Europe, même si l'incidence en Grande-Bretagne semble un peu supérieure[3].
Le sujet moyen a environ 80 ans. Les deux sexes, et toutes les origines ethniques y semblent également vulnérables.
De rares cas ont été constatés chez l'enfant (une cinquantaine de cas dans la littérature, parfois à la suite d'une vaccination (ex contre l'hépatite B[4]) ou à la suite d'une médication (diurétique de l'anse[5]). Chez l'enfant, la maladie survient la première année de vie, avec des lésions bulleuses sur une peau érythémateuse ou sur une peau normale. Les pemphigoïdes bulleuses de l'enfant sont classés en deux sous-types[6] : pemphigoïde bulleuse infantile (PBI) et pemphigoïde bulleuse infantile localisée à la vulve. Chez l'enfant les symptômes disparaissent d'autant plus vite que le traitement est commencé précocement[6].
La maladie est très fréquemment associée avec des atteintes neurologiques[7] (maladie de Parkinson, épilepsie, sclérose en plaques ou troubles cognitifs) ainsi qu'avec le psoriasis[8].
Rarement, d'autres troubles immunitaires sont associés (polyarthrite rhumatoïde, lupus érythémateux systémique, cirrhose biliaire primaire, pemphigus vulgaire, etc.), il peut alors s'agir d'une « maladie auto-immune multiple » ou d'une association fortuite[9]. Le diabète et le psoriasis sont plus souvent associés[9]. Il ne semble pas y avoir de risque supplémentaire de cancer (à âge égal)[9].
Description
L'éruption bulleuse est précédée de plusieurs semaines par un prurit avec des plaques d'allure urticarienne ou eczématiforme.
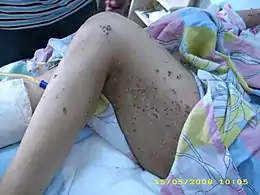
Les bulles ou cloques sont souvent épaisses de plusieurs millimètres, contiennent de la lymphe claire, et sont visuellement assez semblables aux cloques qui apparaissent lors de certaines brûlures. Elles apparaissent principalement sur les membres (jusque sous la plante du pied parfois) et sont accompagnées d'un prurit intense. L'importance de l'éruption peut être quantifiée par un score[10].
L'atteinte de la muqueuse buccale est possible dans moins d'un cas sur cinq. Les atteintes des autres muqueuses sont beaucoup plus rares[3].
Formes de pemphigoïdes bulleuses
La forme « bulleuse » généralisée est la plus fréquente.
La forme vésiculeuse est plus rare, caractérisée par de très petites cloques, souvent sur les mains avec démangeaisons importantes.
La forme urticarienne comporte un prurit important.
La forme de type prurigo est nodulaire (érythrodermie), sans bulles, à croûtes arrondies évoquant des croûtes de grattage, accompagnées de démangeaisons diffuses et fortes (avec forte gêne le jour et insomnies la nuit).
Il existe des formes localisées.
Symptômes
Un prurit précédant ou accompagnant l'apparition de grandes plaques rouges et de nombreuses bulles dépassant souvent 3 à 4 mm d'épaisseur et de grande taille sont les principaux symptômes externes. Certaines des bulles apparaissant parfois sur de la peau apparemment saine, avec prurit. Les lésions sont généralement symétriques sur les deux moitiés du corps ; essentiellement sur le tronc et les membres ou le cuir chevelu, rarement sur le visage, et exceptionnellement sur ou dans la bouche (aphtes ne guérissant pas et gingivite)[11].
Diagnostic
La biopsie cutanée (sous anesthésie locale) confirme au microscope la position des bulles, entre derme et épiderme.
L'immunofluorescence cutanée directe (sur échantillon de peau du malade) confirme la présence des anticorps à la jonction derme-épiderme.
L'immunofluorescence indirecte utilise de la peau saine, clivée et mise au contact du sérum du patient suspecté : les anticorps présents se fixent alors sur la jonction derme-épiderme et sont détectés par immunofluorescence[12]. Le test ELISA permette d'évaluer la présence d'anticorps spécifiques directement dans le sérum du patient. La spécificité de ce dernier test est excellente mais est négatif dans environ 10 % des cas avérés[13].
Le signe de Nikolsky est absent (pas de décollement de la peau aux frottements).
Évolution et pronostic
Des surinfections sont possibles, qu'on prévient par des désinfectants (antiseptiques en applications locales et quotidiennes).
La peau ne jouant plus son rôle normal de protection contre la déshydratation, le patient doit boire en suffisance.
la maladie évolue en poussées successives. Sauf grattage intense, les bulles ne laissent pas de cicatrice atrophiques (plissées et saillantes), mais parfois des taches colorées ou de petits kystes durs. Sans traitement adapté, des lésions étendues peuvent conduire à la déshydratation, à une déprotéinisation, dangereuses pour les personnes très âgées (et nécessitant une bonne alimentation, régulière et non-carencée en protéines). La présence d'un taux élevé d'anticorps anti PB180 à l'arrêt du traitement indiquerait un risque important de récidive[14]. L'âge et un score de Karnofsky de 40 ou moins pourraient aussi indiquer un risque de mortalité plus élevé[15]. Des comorbidités et la tendance à utiliser des immunosuppresseurs et/ou des corticoïdes systémiques influent sans doute aussi sur la morbidité et la mortalité globale.
La corticothérapie au long cours, traitement de la maladie, a des effets secondaires. Ils sont plus marqués chez les personnes âgées.
Il s'agit d'une maladie pouvant être grave. En France, selon les données disponibles au début des années 2000, le taux de mortalité dépassait 30 % après un an de traitement[15], soit six fois plus que la mortalité d'une population du même âge, indemne de l'affection[2]. Les facteurs de mauvais pronostics sont l'âge avancé, les doses requises de corticoïdes et le taux bas d'albumine dans le sang[16]. Le pronostic est plus favorable pour les enfants.
La guérison — en l'absence de complications — est effective dans un intervalle d'un à cinq ans. À distance, une association entre la maladie et la survenue tardive d'une maladie de Parkinson ou d'Alzheimer[17] ou d'une maladie cérébrovasculaire[7] est décrite. De même, le risque de survenue d'un cancer semble augmenter[18].
Confusions possibles
Le diagnostic différentiel doit porter sur les autres maladies bulleuses auto-immunes (plus rares, dont pemphigoïde des muqueuses ou épidermolyse bulleuse acquise), les autres causes d'éruptions bulleuses (urticaire, gale…).
Des tests de type immunotransfert ou immunomicroscopie électronique confirmeront ou infirmeront le diagnostic, et d'adapter le traitement (qui diffère, de même que les besoins de surveillance pour la pemphigoïde cicatricielle ou l'épidermolyse bulleuse acquise.
Causes
Les causes de cette maladie sont encore mal comprises. Comme toutes les maladies auto-immunes, elle implique que les défenses immunitaires, au lieu de n'attaquer que des éléments « étrangers » à l'organisme (bactéries, virus…), se retournent ses propres cellules en produisant des auto-anticorps, qui conduisent à une autodestruction de certains tissus (os, articulations, peau, vaisseaux, etc. devenus auto-antigènes[19]), avec des réactions inflammatoires intenses.
Selon les cas, des facteurs environnementaux, hormonaux et/ou génétiques sont suspectés bien qu'il ne s'agisse pas d'une maladie héréditaire (Quelques formes familiales, très rares ont été décrites, mais un seul de deux vrais jumeaux peut développer la maladie, on parle donc de simple prédisposition génétique, en supposant que d'autres causes ne soient pas passées inaperçues).
Dans quelques cas, des médicaments (spironolactone, bumétanide, fluoxétine, anticancéreux de type inhibiteurs de PD-1[20], etc.) pourraient avoir été des éléments déclencheurs de pemphigoïde bulleuse, de même qu'une photochimiothérapie (puvathérapie associant psoralène et rayonnement UV-A)[3]. Le rôle potentiel de déclencheur de la spironolactone et, à un moindre degré, de neuroleptiques est suspecté[21]. Un cas a été décrit après un vaccin antigrippal[22].
Les auto-anticorps ciblent deux protéines de structure de la peau :
- AgPB230 (ou BPAG1[9]), protéine strictement intracellulaire de masse moléculaire de 230 kDa[9] ;
- AgPB180 (aussi nommée BPAG2 ou collagène XVII, protéine transmembranaire ayant une large extension extracellulaire, de masse moléculaire de 180 kDa[9] - [19]. le caractère pathologique de l'auto-anticorps ciblant cette protéine a été démontré chez la souris[23], alors qu'il est moins évident pour l'AgPB230 qui est théoriquement protégé du contact avec son auto-anticorps.
Ces 2 protéines sont situées dans les hémidesmosomes, à l'interface entre derme et épiderme. C'est là que se forment les cloques. Ils sont également présent dans le système nerveux central, ce qui pourrait expliquer la proportion importante de maladies neurologiques chez les porteurs de cette affection[3].
Autres formes
Il existe aussi d'autres formes particulières de dermatoses bulleuses auto-immunes :
- chez la femme enceinte après le premier trimestre, on retrouve la pemphigoïde gestationis caractérisée par l'éruption bulleuse de la région périombilicale, récidivante à chaque grossesse, et à la prise d'hormones œstroprogestatives (pilule contraceptive) ;
- chez l'enfant et l'adulte jeune, la dermatite herpétiforme se caractérise par l'éruption bulleuse des zones cutanées postérieures (dos, fesses, face d'extension des bras) accompagnée d'un prurit. Le traitement utilise le Disulone et un régime sans gluten peut-être proposé en cas de maladie cœliaque.
Traitement
Dans les cas graves (formes généralisées ou étendues), une hospitalisation initiale est nécessaire.
Deux types de traitement sont possibles[24] :
- monothérapie en corticothérapie systémique (prednisone: 1 mg/kg/jour en général) qui était le traitement le plus fréquent dans les années 1990[25], surtout dans les cas de pemphigoïdes bulleuses généralisées[26], avec des effets secondaires parfois importants ;
- corticothérapie locale en pommade (application sur tout le corps, répétées chaque jour durant 1 an à 5 ans), recommandée en France, à condition de commencer par un traitement d'induction à doses élevées (propionate de clobétasol à dose de 30 à 40 g/jour pour l'adulte, durant un mois sauf si les bulles ont déjà disparu. Ensuite la dose est progressivement diminuée pour atteindre un traitement d'entretien à (5-7 mg/jour de prednisone ou 20-30 g/semaine de propionate de clobétasol)[9]. Ce traitement est plus contraignant et plus long que la corticothérapie systémique, mais aussi efficace, voire plus, avec des effets secondaires moindres et moins dangereux que par voie orale[27] - [28].
Quand les bulles n'apparaissent plus et que le prurit a disparu, le traitement perdure un mois environ, puis est — en 4 à 6 mois — peu à peu diminué et stoppé jusqu'à une éventuelle récidive. Au total, selon les cas de 1 à 5 ans de traitement auront été nécessaires (et ce délai est allongé en cas de complications).
En cas (très rare) d'inefficacité du traitement (cortico-résistance), le médecin propose des immunosuppresseurs (méthotrexate, azathioprine), qui limiteront les doses de corticoïdes nécessaires en cas de rechutes. Le rituximab ou l'omalizumab constituent également des alternatives thérapeutiques[29].
La doxycycline pourrait constituer une alternative aux corticoïdes[30].
Dans les formes graves, un soutien psychologique est utile et parfois nécessaire.
Perspective, recherche médicale
En France, deux hôpitaux sont centres de références[11] labellisés et il existe un groupe de recherche visant à mieux comprendre cette maladie.
Notes et références
- Orphanet Encyclopédie, [www.orpha.net/data/patho/Pub/fr/PemphigoideBulleuse-FRfrPub8663v01.pdf La pemphigoïde bulleuse], septembre 2008 [PDF]
- (en) Joly P, Baricault S, Sparsa A et al. « Incidence and mortality of bullous pemphigoid in France » J Invest Dermatol. 2012;132:1998-2004
- (en) Schmidt E, Zillikens D, « Pemphigoid diseases » Lancet 2013;381:320-332
- (en) Erbagci Z. « Childhood bullous pemphigoid following hepatitis B immunization » J Dermatol. 2002;29:781-5.
- Lloyd-Lavery A, Chi CC, Wojnarowska F, and Taghipour K, The associations between bullous pemphigoid and drug use: a UK case-control study, JAMA Dermatol, 2013;149:58–62
- (en) Fisler RE, Saeb M, Liang MG, Howard RM, McKee PH. « Childhood bullous pemphigoid: a clinicopathologic study and review of the literature » Am J Dermatopathol. 2003; 25:183-9.
- (en) Taghipour K, Chi CC, Vincent A, Groves RW, Venning V, Wojnarowska F. « The association of bullous pemphigoid with cerebrovascular disease and dementia: a case-control study » Arch Dermatol. 2010;146:1251-1254
- (en) Chen YJ, Wu CY, Lin MW et al. « Comorbidity profiles among patients with bullous pemphigoid: a nationwide population-based study » Br J Dermatol. 2011;165:593-599
- (en) Orphanet/ Pr Philippe Bernard, Dr Enrico Bertini, « Bullous pemphigoid » juin 2001, mis à jour en 2003 puis avril 2006 [PDF]
- (en) Murrell DF, Daniel BS, Joly P et al. « Definitions and outcome measures for bullous pemphigoid: recommendations by an international panel of experts » J Am Acad Dermatol. 2011;66:479-485
- Association Pemphigus - Pemphigoïde France, Signes et symptômes
- (en) Gammon WR, Briggaman RA, Inman AO, Queen LL, Wheeler CE. « Differentiating anti-lamina lucida and anti-sublamina densa anti-BMZ antibodies by indirect immunofluorescence on 1·0 M sodium chloride-separated skin » J Invest Dermatol. 1984;82:139-144
- (en) Sitaru C, Dahnrich C, Probst C et al. « Enzyme-linked immunosorbent assay using multimers of the 16th non-collagenous domain of the BP180 antigen for sensitive and specific detection of pemphigoid autoantibodies » Exp Dermatol. 2007;16:770-777
- (en) Bernard P, Reguiai Z, Tancrede-Bohin E et al. « Risk factors for relapse in patients with bullous pemphigoid in clinical remission: a multicenter, prospective, cohort study » Arch Dermatol. 2009;145:537-542
- (en) Joly P, Benichou J, Lok C et al. « Prediction of survival for patients with bullous pemphigoid » Arch Dermatol. 2005;141:691-8.
- (en) Rzany B, Partscht K, Jung M et al. « Risk factors for lethal outcome in patients with bullous pemphigoid: low serum albumin level, high dosage of glucocorticosteroids, and old age » Arch Dermatol. 2002;138:903-908
- Brick KE, Weaver CH, Savica R et al. A population-based study of the association between bullous pemphigoid and neurologic disorders, J Am Acad Dermatol, 2014; 71: 1191–1197
- Schulze F, Neumann K, Recke A, Zillikens D, Linder R, Schmidt E, Malignancies in pemphigus and pemphigoid diseases, J Invest Dermatol, 2015;135:1445–1447
- (en) Guidice GJ, Emery DJ, Diaz LA. « Cloning and primary structural analysis of the bullous pemphigoid autoantigen BP180 » J Invest Dermatol. 1992; 99:243-50
- Lopez AT, Khanna T, AntonovN, Audrey-Bayan C, Geskin L. A review of bullous pemphigoid associated with PD-1 and PD-L1 inhibitors, Int J Dermatol, 2018;57:664-669
- (en) Bastuji-Garin S, Joly P, Picard-Dahan C, Bernard P, Vaillant L, Pauwels C, Salagnac V, Lok C, Roujeau JC. « Drugs associated with bullous pemphigoid : a case-control study » Arch Dermatol. 1996; 132:272-6.
- (en) Walmsley N, Hampton P, « Bullous pemphigoid triggered by swine flu vaccination: case report and review of vaccine triggered pemphigoid » J Dermatol Case Rep. 2011;5:74-76
- (en) Liu Z, Diaz LA, Troy JL et al. « A passive transfer model of the organ-specific auto-immune disease: bullous pemphigoid, using antibodies generated against the hemidesmosomal antigen BP180 » J Clin Invest. 1993;92:2480-8.
- (en) Joly P, Roujeau JC, Benichou J, Picard C, Dreno B, Delaporte E, Vaillant L, D'Incan M, Plantin P, Bedane C, Young P, Bernard P. « Bullous Diseases French Study Group. A comparison of oral and topical corticosteroids in patients with bullous pemphigoid » N Engl J Med. 2002; 31;346:321-7.
- (en) Fine JD. « Management of autoimmune bullous disases » N Engl J Med. 1995; 333:1475-84.
- (en) Mutassim DF. « How to diagnose and manage autoimmune disease » Skin & Aging 1999;7(8):59-63.
- (en) Joly P, Roujeau JC, Benichou J et al. « A comparison of oral and topical corticosteroids in patients with bullous pemphigoid » N Engl J Med. 2002;346:321-327
- (en) Kirtschig G, Middleton P, Bennett C, Murrell DF, Wojnarowska F, Khumalo NP, « Interventions for bullous pemphigoid » Cochrane Database Syst Rev. 2010;10.CD002292
- Kremer N, Snast I, Cohen ES et al. Rituximab and omalizumab for the treatment of bullous pemphigoid: a systematic review of the literature, Am J Clin Dermatol, 2019;20:209-216
- Williams HC, Wojnarowska F, Kirtschig G et al. Doxycycline versus prednisolone as an initial treatment strategy for bullous pemphigoid: a pragmatic, non-inferiority, randomised controlled trial, Lancet, 2017;389:1630–1638