Propriété colligative
En chimie physique une propriété colligative d'une solution chimique correspond à la différence entre une propriété donnée d'un solvant pur liquide et la même propriété de ce solvant en présence d'un soluté. Le mot colligatif vient du mot latin colligatus qui signifie lié ensemble, une propriété colligative étant la conséquence de la dilution du solvant par le soluté.
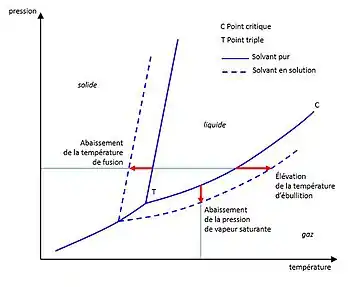
Les propriétés colligatives sont surtout étudiées sur les solutions liquides dans lesquelles le soluté est très fortement dilué et qui peuvent en conséquence être considérées comme des solutions idéales : elle ne dépendent alors que de la quantité (nombre de moles) du soluté et non de sa nature chimique. La condition de solution idéale permet en effet de négliger les interactions particulières existant entre solvant et soluté et qui diffèrent selon les espèces chimiques présentes.
Sous la condition de la solution idéale, les propriétés colligatives fournissent des moyens expérimentaux de détermination de la masse molaire du soluté en ne connaissant que les propriétés du solvant pur. Elles permettent également d'étudier le comportement du soluté en solution, celui-ci pouvant en effet se dissocier (par exemple en formant des ions) ou s'associer (par exemple en formant des dimères).
Les propriétés colligatives comprennent :
- l'abaissement de la pression de vapeur saturante du solvant (loi de la tonométrie) ;
- l'élévation de la température d'ébullition du solvant (loi de l'ébulliométrie) ;
- l'abaissement de la température de fusion (ou température de solidification ou température de congélation) du solvant (loi de la cryométrie) ;
- la pression osmotique (loi de l'osmométrie).
Les trois premières lois ont été énoncées par François-Marie Raoult à partir de 1878, elles sont appelées collectivement lois de Raoult. La quatrième loi a été énoncée par Jacobus Henricus van 't Hoff en 1886, ce qui le valut le premier prix Nobel de chimie en 1901.
Principe général
Diminution du potentiel chimique du solvant
Lorsqu'un solvant pur liquide est en équilibre avec une autre phase (solvant pur gazeux ou solide) sous la pression et à la température , les potentiels chimiques de ce corps dans les deux phases sont égaux :




avec :
- le potentiel chimique du solvant pur en phase ;
- le potentiel chimique du solvant pur en phase liquide.


Lorsque l'on introduit une quantité très petite de soluté dans le solvant liquide, la solution obtenue peut être considérée comme idéale. Le potentiel chimique du solvant en phase liquide est alors modifié selon :

avec :
- le potentiel chimique du solvant dans la solution liquide ;
- la constante universelle des gaz parfaits ;
- la fraction molaire du solvant dans la solution liquide.



Puisque et sont positives et que , alors :


À pression et température constantes la présence du soluté diminue le potentiel chimique du solvant en phase liquide. En conséquence, lors de la formation d'une solution les molécules du solvant ont moins tendance à se déplacer vers une autre phase, les solutions liquides deviennent ainsi stables :
- à des températures supérieures à la température d'ébullition du solvant pur : la température d'ébullition augmente ;
- à des températures inférieures à la température de fusion du solvant pur : la température de fusion diminue ;
- à des pressions inférieures à la pression de vapeur saturante du solvant pur : la pression de vapeur saturante diminue.
Rétablissement d'un équilibre de phases
-f(T).png.webp)
On considère que le soluté est absent de la phase (le soluté n'est pas volatil dans le cas d'une phase gazeuse, ou ne se solidifie pas dans le cas d'une phase solide). La phase est donc exclusivement constituée du solvant pur (à l'état gazeux, liquide ou solide).
L'équilibre de phases du solvant en présence du soluté ne peut pas s'établir à et , puisque . Les propriétés colligatives correspondent aux modifications de pression ou de température nécessaires pour retrouver un équilibre de phases avec, toujours dans l'hypothèse d'une solution liquide suffisamment diluée pour être idéale :


- Modification de la pression
- Si l'on travaille à température constante, on ne peut retrouver un équilibre de phases du solvant qu'en modifiant la pression afin d'obtenir :

- Si la phase est une phase gazeuse, alors : la pression de vapeur saturante du solvant est abaissée.

- Cas de l'osmose
- Dans le cas particulier de l'osmose, la phase est le solvant pur à l'état liquide. Une membrane semi-perméable sépare le solvant liquide pur de la solution. Cette membrane permet d'établir un équilibre mécanique dans lequel les deux phases liquides sont sous des pressions différentes à la même température. On peut ainsi conserver la pression dans le solvant pur et modifier la pression uniquement dans la solution :

- avec : la présence du soluté augmente la pression d'équilibre de la solution, il s'agit de la pression osmotique.

- Modification de la température
- Si l'on travaille à pression constante, on ne peut retrouver un équilibre de phases du solvant qu'en modifiant la température afin d'obtenir :

- Si la phase est une phase gazeuse, alors : la température d'ébullition du solvant est élevée.
- Si la phase est une phase solide, alors : la température de fusion du solvant est abaissée.


Énoncé des lois
Les écarts colligatifs sont tous positifs. La propriété de la solution est calculée pour un abaissement par soustraction de l'écart colligatif à la propriété du solvant pur : ; pour une élévation l'écart colligatif est ajouté à la propriété du solvant pur : .





Abaissement de la pression de vapeur saturante
Lorsque l'on considère un solvant contenant un soluté , la pression de vapeur saturante du solvant avec le soluté est plus basse que la pression de vapeur saturante du solvant seul à la même température. La loi de la tonométrie s'énonce ainsi :


« Dans une solution binaire, l'abaissement relatif de la pression de vapeur saturante du solvant est égal à la fraction molaire du soluté. »
Soit :
| Loi de la tonométrie : |

avec :
- la pression de vapeur saturante du solvant pur ;
- l'abaissement absolu de la pression de vapeur saturante du solvant en présence du soluté ;
- la fraction molaire du soluté.



Le terme est l'abaissement relatif de la pression de vapeur saturante du solvant.

Autrement dit, à température constante, la pression de vapeur saturante du solvant pur passe à en présence d'un soluté. La fraction molaire du soluté étant une grandeur positive, l'abaissement de pression est positif. Ainsi l'ajout d'un soluté fait-il diminuer la pression de vapeur saturante du solvant à température constante (, soit ).



La loi de la tonométrie peut également s'exprimer en fonction de la molalité du soluté (en mol/kg) selon :


avec :
- la quantité du soluté (en mol) ;
- la masse du solvant en solution (en kg) ;
- la masse molaire du solvant (en g/mol).



Élévation de la température d'ébullition
Lorsque l'on considère un solvant contenant un soluté , la température d'ébullition du solvant avec le soluté est plus haute que la température d'ébullition du solvant seul. La loi de l'ébulliométrie s'énonce ainsi :
« L'élévation de la température d'ébullition est proportionnelle à la fraction molaire du soluté. »
Soit (en remarquant que pour un corps pur la température d'ébullition est égale à la température de vaporisation) :
| Loi de l'ébulliométrie : |

avec :
- l'élévation de la température d'ébullition du solvant (en K) ;
- la constante ébullioscopique du solvant (en K) ;
- la fraction molaire du soluté (en mol/mol).


La constante ébullioscopique ne dépend que des propriétés du solvant :

avec :
- la constante universelle des gaz parfaits (en J/(K·mol)) ;
- la température d'ébullition du solvant pur (en K) ;
- l'enthalpie de vaporisation du solvant pur à (en J/mol).


Sous cette forme, la constante ébullioscopique a la dimension d'une température, elle s'exprime en kelvins (K).
Autrement dit, à pression constante, la température d'ébullition du solvant pur passe à en présence d'un soluté. L'enthalpie de vaporisation étant une grandeur positive, la constante ébullioscopique est positive. Ainsi l'ajout d'un soluté fait-il augmenter la température d'ébullition du solvant à pression constante (, soit ).



La loi de l'ébulliométrie peut également s'exprimer en fonction de la molalité du soluté (en mol/kg) selon :


avec :
- la quantité du soluté (en mol) ;
- la masse du solvant en solution (en kg) ;
- la masse molaire du solvant (en g/mol).
Sous cette forme, la constante ébullioscopique s'exprime en K·kg/mol (ou en °C·kg/mol), elle ne dépend toujours que des propriétés du solvant pur.
Abaissement de la température de fusion
Lorsque l'on considère un solvant contenant un soluté , la température de solidification du solvant avec le soluté est plus basse que la température de solidification du solvant seul. La loi de la cryométrie s'énonce ainsi :
« L'abaissement de la température de solidification est proportionnel à la fraction molaire du soluté. »
Soit (en remarquant que pour un corps pur la température de solidification - ou température de congélation - est égale à la température de fusion) :
| Loi de la cryométrie : |

avec :
- l'abaissement de la température de fusion du solvant (en K) ;
- la constante cryoscopique du solvant (en K) ;
- la fraction molaire du soluté (en mol/mol).


La constante cryoscopique ne dépend que des propriétés du solvant :

avec :
- la constante universelle des gaz parfaits (en J/(K·mol)) ;
- la température de fusion du solvant pur (en K) ;
- l'enthalpie de fusion du solvant pur à (en J/mol).


Sous cette forme, la constante cryoscopique a la dimension d'une température, elle s'exprime en kelvins (K).
Autrement dit, à pression constante, la température de fusion du solvant pur passe à en présence d'un soluté. L'enthalpie de fusion étant une grandeur positive, la constante cryoscopique est positive. Ainsi l'ajout d'un soluté fait-il diminuer la température de fusion du solvant à pression constante (, soit ).



La loi de la cryométrie peut également s'exprimer en fonction de la molalité du soluté (en mol/kg) selon :


avec :
- la quantité du soluté (en mol) ;
- la masse du solvant en solution (en kg) ;
- la masse molaire du solvant (en g/mol).
Sous cette forme, la constante cryoscopique s'exprime en K·kg/mol (ou en °C·kg/mol), elle ne dépend toujours que des propriétés du solvant pur.
Pression osmotique

Lorsque l'on place un solvant pur et une solution d'un soluté quelconque dans le même solvant de part et d'autre d'une membrane semi-perméable (ne laissant passer que le solvant), le solvant migre spontanément à travers la membrane du compartiment A contenant le solvant pur vers le compartiment B contenant la solution : ce phénomène est appelé osmose. Au bout d'un certain temps la migration du solvant cesse et un équilibre s'établit entre les deux compartiments. À l'équilibre osmotique la membrane subit une pression plus importante de la part de la solution que de la part du solvant pur ; le solvant migre donc du compartiment de plus faible pression, le compartiment A, vers celui de plus forte pression, le compartiment B.
La loi de van 't Hoff permet de calculer le surcroît de pression exercée par le compartiment contenant la solution dans le cas des solutions très diluées selon :
| Loi de van 't Hoff, ou loi de l'osmométrie : |

avec :
- la pression osmotique (en Pa), c'est-à-dire le surcroît de pression exercée sur la membrane par la solution du compartiment B par rapport au solvant pur du compartiment A ;
- le volume de la solution du compartiment B (en m3) ;
- la quantité (ou nombre de moles) de soluté en solution (en mol) ;
- la constante universelle des gaz parfaits (en J/(K·mol)) ;
- la température (en K).



La forme de cette loi rappelle celle des gaz parfaits . Elle est totalement indépendante des propriétés intrinsèques du solvant et du soluté. Quelles que soient les conditions opératoires , c'est donc toujours le compartiment B contenant la solution qui exerce la pression la plus importante sur la membrane.


La loi de l'osmométrie peut également s'exprimer en fonction de la molalité du soluté (en mol/kg) selon :

avec :
- la quantité du soluté (en mol) ;
- la masse du solvant en solution (en kg) ;
- la masse volumique du solvant pur à la température (en kg/m3).

Applications
Détermination d'une masse molaire
Les propriétés colligatives permettent notamment de déterminer la masse molaire d'un soluté.

De façon générale, on crée une solution en dissolvant une masse de soluté dans un solvant liquide. Cette masse doit être suffisamment faible afin que la solution obtenue soit le plus proche possible d'une solution idéale. On détermine expérimentalement l'un des écarts colligatifs , , ou de cette solution. Les lois énoncées précédemment permettent de calculer la quantité du soluté en solution. La masse molaire du soluté peut donc être obtenue selon : .


- Exemple[1]
- On prépare une solution de 1,25 g de salicylate de méthyle (essence de gaulthérie couchée ou wintergreen) dans 99,0 g de benzène. La température d'ébullition du benzène pur est de 80,1 °C, celle de la solution de 80,31 °C. La constante ébullioscopique molale du benzène vaut 2,53 °C·kg/mol.

- L'élévation de la température d'ébullition est de :

- Le nombre de moles du salicylate de méthyle est de :

- La masse molaire du salicylate de méthyle est donc de :

Étude du comportement d'un soluté en solution
Dans certains cas, notamment celui des électrolytes, les écarts colligatifs , , ou obtenus expérimentalement sont plus importants que ceux prévus théoriquement. Ceci s'explique par la dissociation (notamment sous forme d'ions) du soluté dans la solution : le nombre de particules en solution est plus élevé que prévu. Ainsi, le chlorure de sodium (sel de table) NaCl se dissocie dans l'eau en deux ions selon :

L'abaissement de la température de fusion d'une solution aqueuse de sel est quasiment deux fois plus important que prévu. Le sulfate de sodium Na2SO4 se dissocie dans l'eau en trois ions selon :

L'abaissement de la température de fusion d'une solution aqueuse de sulfate de sodium est quasiment trois fois plus important que prévu[2].
Dans d'autres cas les écarts colligatifs expérimentaux sont inférieurs aux écarts prévus théoriquement. Ceci s'explique par une association des molécules de soluté en solution : le nombre de particules en solution est plus petit que prévu. Ainsi, dans le benzène l'acide acétique forme des dimères selon :

En conséquence, les écarts colligatifs de ces solutions sont la moitié de ceux attendus théoriquement.
De façon générale, les quatre lois relatives aux propriétés colligatives peuvent s'écrire dans leur forme molale selon :

avec :
- l'écart colligatif ;
- une constante ne dépendant que des propriétés du solvant ;
- la masse de solvant ;
- la quantité de soluté.


On définit :
- l'écart colligatif théorique que l'on devrait obtenir si le soluté ne se dissociait et ne s'associait pas ;
- l'écart colligatif que l'on obtient expérimentalement ;
- la quantité de soluté théorique, ou apparente, en l'absence de toute dissociation ou association ;
- la quantité de soluté réelle, conséquence d'une dissociation ou d'une association.




On a :


Le facteur de van 't Hoff est défini par :


Ce facteur permet de déterminer le nombre de particules produites par une particule de soluté en solution :

Une particule de soluté produit particules en solution. Si le soluté ne subit aucune modification lors de sa mise en solution, alors ; si le soluté se dissocie dans la solution, alors ; si le soluté s'associe dans la solution, alors .



Exemple 1 - dissociation d'un sel[3]
- La température de fusion d'une solution aqueuse de [Co(NH3)(NO2)]Cl à une molalité de 0,002 mol/kg est de −0,007 32 °C. La constante cryoscopique molale de l'eau vaut 1,86 °C·kg/mol.

- L'abaissement de la température de fusion théorique est de :

- La température de fusion de l'eau pure est de 0 °C. L'abaissement de la température de fusion expérimental est de :

- Le facteur de van 't Hoff est donc de :

- Le corps dissout se dissocie donc en deux ions. D'autres expériences montrent qu'il s'agit de [Co(NH3)(NO2)]+ et Cl−.
Exemple 2 - association du soufre[4]
- 0,24 g de soufre S, de masse atomique 32 g/mol, dissout dans 100 g de tétrachlorure de carbone CCl4 abaisse la température de fusion de celui-ci de 0,28 °C. La constante cryoscopique molale du CCl4 vaut 30 °C·kg/mol.
- La molalité du soufre est de :

- L'abaissement théorique de la température de fusion du CCl4 est de :

- Le facteur de van 't Hoff est donc de :

- Le soufre dissout se présente sous une forme moléculaire comprenant huit atomes : le cyclooctasoufre de formule S8.
Notes et références
Notes
- Kotz et al., p. 30.
- Kotz et al., p. 32.
- Kotz et al., p. 33.
- Atkins et al., p. 394.
Bibliographie
- Détermination des poids moléculaires : mémoires de MM. Avogadro, Ampère, Raoult, van 't Hoff, D. Berthelot, Gauthier-Villars, (lire en ligne) (sur Gallica).
- Paul Arnaud, Françoise Rouquérol, Gilberte Chambaud, Roland Lissillour, Abdou Boucekkine, Renaud Bouchet, Florence Boulc'h et Virginie Hornebecq, Chimie générale : Cours avec 330 questions et exercices corrigés et 200 QCM, Dunod, coll. « Les cours de Paul Arnaud », , 8e éd., 672 p. (ISBN 978-2-10-074482-4, lire en ligne), p. 337-341.
- Peter William Atkins, Loretta Jones et Leroy Laverman (trad. de l'anglais), Principes de chimie, Louvain-la-Neuve, De Boeck Superieur, , 4e éd., 1088 p. (ISBN 978-2-8073-0638-7, lire en ligne), p. 388-398.
- Peter William Atkins et Julio De Paula, Chimie Physique, De Boeck Superieur, , 4e éd., 1024 p. (ISBN 9782804166519, lire en ligne), p. 167-175.
- Mohamed Ayadim et Jean-Louis Habib Jiwan, Chimie générale, Louvain, Presses universitaires de Louvain, coll. « Cours universitaires », , 376 p. (ISBN 978-2-87558-214-0, lire en ligne), p. 260-266.
- Danielle Baeyens-Volant, Pascal Laurent et Nathalie Warzée, Chimie des solutions : Exercices et méthodes, Dunod, coll. « Chimie générale », , 320 p. (ISBN 978-2-10-076593-5, lire en ligne), p. 30-36.
- John C. Kotz et Paul M. Treichel Jr (trad. de l'anglais), Chimie des solutions, Bruxelles/Issy-les-Moulineaux, De Boeck Supérieur, coll. « Chimie générale », , 358 p. (ISBN 978-2-8041-5232-1, lire en ligne), p. 23-36.
- Claude Strazielle, Caractérisation par la détermination des masses moléculaires, vol. PE 595, Éditions techniques de l'ingénieur, (lire en ligne), p. 1-4.