Champ de force (chimie)
Dans le cadre de la mécanique moléculaire, un champ de force est un ensemble de potentiels et de paramètres permettant de décrire la structure de l'énergie potentielle d'un système de particules (typiquement, des atomes, mais non exclusivement). L'usage de l'expression champ de force en chimie et biologie numériques diffère ainsi de celui de la physique, où il indique en général un gradient négatif d'un potentiel scalaire. Les potentiels et paramètres d'un champ de force donné sont déterminés à partir de résultats expérimentaux et de calculs précis en mécanique quantique.
Il existe, en fonction des systèmes étudiés, de nombreux champs de force différents. Les champs de force tout-atome (all atom) donnent des paramètres pour tous les atomes du système, y compris l'hydrogène, alors que les champs de force atomes unifiés (unified atom) ne prennent pas en compte les atomes d'hydrogène sauf si ces derniers sont polaires (H est traité comme un centre d'interaction). Les champs de force basés sur les atomes lourds ne prennent pas du tout en compte les atomes d'hydrogène. Certains champs de force sont basés sur des modèles simplifiés de tout ou partie d'un ou plusieurs objets comme les champs à gros grains (coarse grained) fréquemment utilisés dans les simulations longues de protéines, ou les champs basés sur des grilles et réseaux (in lattice).
Le choix d'un champ, spécifique d'un système et plus ou moins efficace, dépend des études que l'on doit produire et des propriétés recherchées.
Forme générale
La forme fonctionnelle de base d'un champ de force comprend les termes de liaisons reliés aux atomes liés par des liaisons covalentes, et les termes d'interactions (termes dit non-liés ou non-covalents) qui décrivent les interactions à longue portée (électrostatique et de Van der Waals). La décomposition spécifique en chacun de ces termes dépend du champ de force, mais la forme générale de l'énergie totale dans un champ de force additif peut être écrite :

où les composants des contributions covalentes et non-covalentes sont données par les sommes suivantes :


Les termes de liaison et d'angle sont habituellement modélisés comme des oscillateurs harmoniques dans les champs de force ne permettant pas la formation/destruction de liaison. Une description plus réaliste d'une liaison covalente soumise à un étirement important peut être donnée par un potentiel de Morse, plus couteux. La forme fonctionnelle du reste des termes liés est très variable. Des potentiels dièdres sont également habituellement inclus. De plus, des termes de « torsion impropre » peuvent être ajoutés pour forcer la planéité des cycles aromatiques et autres systèmes conjugués, ainsi que des termes « croisés » décrivant le couplage entre les différentes variables internes, comme les angles et les longueurs de liaison. Certains champs de force incluent des termes explicites pour les liaisons hydrogène.
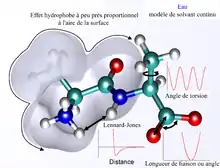
Les termes non-liés sont plus couteux en temps de calcul car ils doivent prendre en compte bien plus d'interactions par atome. Un des choix les plus courants est de limiter les interactions aux énergies de paire. Le terme de van der Waals est en général calculé à l'aide d'un potentiel de Lennard-Jones, et le terme électrostatique par la loi de Coulomb, bien que ces deux termes puissent être atténués ou augmentés par un facteur constant afin de tenir compte de la polarisabilité électronique, et produire ainsi un meilleur accord avec l'expérience.
Paramétrage
En plus de la forme fonctionnelle des potentiels, un champ de force définit un ensemble de paramètres pour chacun des types d'atomes. Ainsi par exemple, un champ de force comprendra des paramètres distincts pour un atome d'oxygène dans un groupe carbonyle et dans un groupe hydroxyle. Un ensemble typique de paramètres comprend des valeurs pour la masse atomique, le rayon de van der Waals et la charge partielle d'atomes individuels, et des valeurs d'équilibre de longueurs de liaison, de mesures d'angles plans et dièdres pour des paires, triplets et quadruplets d'atomes liées, et les valeurs de constante de force pour chaque potentiel. Les champs de force les plus courants utilisent un modèle de charge fixe dans lequel chaque atome se voit affecter une charge électrostatique qui n'est pas affectée par l'environnement local. Les développements proposés pour les champs de force de prochaine génération incluent des modèles pour la polarisabilité dans lesquels les charges des particules sont influencées par celles de leurs voisines. La polarisabilité peut, par exemple, être approchée par l'introduction de dipôles induits ; elle peut aussi être représentée par des particules de Drude, ou des sites porteurs de charges virtuels et sans masses rattachés par un potentiel harmonique de type ressort à chaque atome polarisable. L'introduction de la polarisabilité dans les champs de force pour un usage courant est ralentie par le cout important associé au calcul du champ électrique local.
Bien que de nombreuses simulations moléculaires portent sur des macromolécules biologiques comme les protéines, l'ADN ou l'ARN, les paramètres utilisés pour un type d'atomes donné proviennent généralement des observations de molécules organiques, plus facilement appréhendables que cela soit pour des études expérimentales ou pour des calculs quantiques. Des champs de force différents peuvent être issus de types données expérimentales distinctes, comme l'enthalpie de vaporisation (comme dans le champ OPLS), l'enthalpie de sublimation (comme dans le champ CFF), les moments dipolaires, ou des paramètres spectroscopiques variés (CFF).
Les ensembles de paramètres et les formes fonctionnelles sont définies par les développeurs de champs de force afin d'être auto-cohérents. Parce que les formes fonctionnelles des potentiels varient de manière extensive y compris pour des champs de force très proches (ou entre versions successives d'un même champ de force), les paramètres d'un champ de force ne doivent pas être utilisés avec les potentiels définis pour un autre champ.
Défauts et manques
Tous les champs de force sont fondés sur de nombreuses approximations et construits à partir de différents types de données expérimentales. Ils sont en conséquence appelés empiriques. Certains champs ne tiennent habituellement pas compte de la polarisation électronique de l'environnement, ce qui a pour effet de réduire de manière significative les interactions électrostatiques des charges atomiques partielles. Ce problème fut considéré au moyen du développement des « champs de force polarisables »[1] - [2] ou par utilisation d'une constante diélectrique macroscopique. Cependant, l'utilisation d'une seule valeur pour la constante diélectrique est sujette à caution dans les environnements hautement hétérogènes des protéines ou des membranes biologiques, et la nature du diélectrique dépend du modèle utilisé[3].
Les forces de van der Waals sont aussi très fortement dépendantes de leur environnement, car résultant de l'interaction entre dipôles induits et « instantanés » (voir force intermoléculaire). La théorie initiale de Fritz London sur ces forces n'est valable que dans le vide. Une théorie plus générale sur les forces van der Waals dans un milieu dense fut développée par A.D. McLachlan en 1963, pour laquelle la théorie de Fritz-London est considérée comme un cas spécifique[4]. Cette théorie indique que les interactions de van der Waals dans un milieu sont plus faibles que dans le vide et suivent la loi « comme s'il y avait dissolution », ce qui signifie que les atomes de types différents interagissent plus faiblement que les atomes de même type[5]. Cette théorie contraste avec les lois de combinaison ou l'équation de Slater-Kirkwood utilisées pour le développement des champs de force classiques. Les lois de combinaison indiquent que l'énergie d'interaction de deux atomes distincts (comme C et N) est une moyenne des énergies d'interaction des paires d'atomes correspondantes (soit C-C et N-N). Selon la théorie de McLachlan, les interactions entre particules dans un milieu peuvent aller jusqu'à être complètement répulsives, comme observé dans l'hélium liquide[4]. Les conclusions de la théorie de McLachlan sont étayées par des mesures directes des forces d'attraction entre différents matériaux (constante d'Hamaker), comme indiqué par Jacob Israelachvili dans son livre Intermolecular and surface forces. Il y était indiqué que « l'interaction entre hydrocarbures dans l'eau est environ 10 % de celle dans le vide »[4]. De tels effets ne sont pas pris en compte en mécanique moléculaire classique.
Un autre type de critique vient des applications pratiques, comme la détermination de structure protéique. Ainsi, aucun des participants au CASP n'essaya de raffiner ses modèles afin d'éviter « un embarras majeur pour la mécanique moléculaire, c'est-à-dire le fait que la minimisation de l'énergie ou la dynamique moléculaire conduisent généralement à un modèle peu en accord avec la structure expérimentale »[6]. En fait, les champs de force ont été appliqués avec succès dans les déterminations de structures protéiques dans différentes applications en diffractométrie de rayons X et en résonance magnétique nucléaire, par exemple avec le code XPLOR[7]. Cependant, ces déterminations sont fondées sur des contraintes expérimentales, alors que les champs de force sont principalement destinés à supprimer les interférences atomiques. Les résultats des calculs sont pratiquement les mêmes avec des potentiels de sphères durs (par exemple dans le code DYANA, pour des calculs fondés sur les données RMN)[8], ou avec des programmes de raffinement cristallographique qui n'utilisent aucune fonction de l'énergie. Les lacunes des champs de force restent un goulet d'étranglement pour la modélisation par homologie des protéines[9]. Cette situation a donné naissance au développement de fonctions empiriques de notation spécifiques pour l'assemblage de ligands[10], le repliement de protéine[11] - [12] - [13], la construction de protéines numérique[14] - [15] - [16], ou la modélisation des protéines dans les membranes[17].
Il existe également une opinion selon laquelle la mécanique moléculaire peut opérer avec une énergie non pertinente pour le repliement des protéines ou la liaison de ligand[18]. Les paramètres des champs de force typiques permettent de reproduire l'enthalpie de sublimation. Cependant, le repliement des protéines ou la liaison de ligand sont thermodynamiquement très semblables à une cristallisation ou à des transitions liquide-solide, car ces processus constituent un « gel » de molécules mobiles dans un milieu condensé[19] - [20] - [21]. Ainsi, les variations de l'énergie libre durant ces processus sont supposées représenter une combinaison d'une énergie de type enthalpie de fusion, d'une contribution d'entropie conformationnelle et d'énergie libre de solvatation. L'enthalpie de fusion est significativement plus petite que l'enthalpie de sublimation[4]. Par conséquent, les potentiels décrivant le repliement des protéines ou la liaison de ligand doivent être plus faibles que les potentiels en mécanique moléculaire. En fait, les énergies des liaisons hydrogène dans les protéines sont de ~ -1,5 kcal/mol lorsqu'elles sont estimées à partir de l'ingénierie protéique ou des données sur la transition entre hélice alpha et pelote[22] - [23], mais si l'on estime les mêmes quantités à partir des enthalpies de sublimation des cristaux moléculaires, elles sont de l'ordre de -4 à -6 kcal/mol [24]. La profondeur des potentiels de Lennard-Jones modifiés dérivés des données de l'ingénierie protéique sont aussi plus petites que dans un champ de force classique et respectent une loi « comme s'il y avait dissolution », comme indiqué par la théorie de McLachlan[18].
Champs de force « courants »
Différents champs de force ont été construits afin d'être appliqués à différents problèmes.
Le champ MM2 a ainsi été développé par Norman Allinger dans un premier temps pour l'analyse conformationnelle des hydrocarbures et autres petites molécules organiques. Il a été conçu afin de reproduire les géométries « covalentes » d'équilibre aussi précisément que possible. Il comprend un grand ensemble de paramètres continument redéfinis et mis à jour pour différentes classes de composés organiques (MM3 et MM4)[25] - [26] - [27] - [28] - [29].
Le champ CFF a été développé par Wrshel, Lifson et collaborateurs comme une méthode générale d'unification des études sur les énergies, les structures et vibrations des molécules et cristaux moléculaires. Le programme CFF, développé par Levitt et Warshell, est basé sur une représentation cartésienne de tous les atomes, et sert de base à de nombreux programmes de simulations plus importants.
Le champ ECEPP a été développé de manière spécifique pour la modélisation des peptides et protéines. Il utilise des géométries fixés de résidus d'acides aminés afin de simplifier la surface d'énergie potentielle. La minimisation d'énergie est ainsi menée dans l'espace des angles de torsion des protéines. Les champs MM2 et ECEPP incluent tous deux des potentiels pour les liaisons hydrogène et des potentiels de torsion afin de décrire les rotations autour des liaisons simples. ECEPP/3 a été intégré dans Internal Coordinate Mechanics et FANTOM[30], par exemple.
Les champs AMBER, CHARMM et GROMOS ont été développés en premier lieu pour la dynamique moléculaire des macromolécules, bien qu'ils soient communément employés pour la minimisation d'énergie. Les coordonnées de tous les atomes y sont considérées comme des variables libres.
Champs de force classiques
Voici quelques champs de force « classiques » :
- AMBER (Assisted Model Building and Energy Refinement). Très utilisé pour la modélisation des protéines, ADN et ARN.
- CHARMM (Chemistry at HARvard Molecular Mechanics). Développé initialement à Harvard, très utilisé pour les petites molécules ou les macromolécules. CHARMm est sa version commerciale, proposée par Dassault Systemes BIOVIA.
- CVFF également utilisé massivement pour les petites molécules ou les macromolécules.
- COSMOS-NMR. Champ de force hybride QM/MM (quantique/classique) adapté à de nombreux composés inorganiques, organiques ou macromolécules biologiques. Il inclut le calcul semi-empirique des charges atomiques et propriétés de RMN. Il est d'ailleurs optimisé pour la résolution des structures basée sur la RMN et est introduit dans l'ensemble COSMOS de modélisation moléculaire[31].
- GROMACS. Champ de force optimisé pour l'ensemble du même nom.
- GROMOS. Champ de force intégré dans l'ensemble généraliste GROMOS (GROningen MOlecular Simulation package), dédié à la simulation dynamique des systèmes biomoléculaires. Le champ de force GROMOS (version A) a été développé pour être appliqué aux solutions aqueuses ou apolaires de protéines, nucléotides ou sucres. Cependant, une version pour simulation en phase gaz (version B) de molécules isolées est disponible.
- OPLS (Optimized Potential for Liquid Simulations). Champ de force développé par William L. Jorgensen au département de chimie de Yale. Il en existe plusieurs évolutions comme OPLS-AA, OPLS-UA, OPLS-2001, ou OPLS-2005.
- ECEPP/2. Premier champ de force dédié aux molécules polypeptidiques, développé par F.A.Momany, H.A.Scheraga et collaborateurs.
- QCFF/PI. Champ de force dédié aux molécules conjugées[32] - [33].
Champs de force de deuxième génération
- CFF est une famille de champs de force adaptés à une large gamme de composés organiques, comme pour les polymères, les métaux, etc.
- MMFF (Merck Molecular Force Field) développé pour une large gamme de molécules.
- MM2, MM3, MM4, développés par Norman Allinger, paramétrée pour une large gamme de molécules.
- QVBMM développé par Vernon G.S. Box, paramétré pour traiter les biomolécules et une large gamme de molécules organiques, et inclus dans STR3DI32.
Champs de force polarisables basés sur des dipôles induits
Champs de force polarisables basés sur des charges ponctuelles
- PFF (Polarizable Force Field) développé par Richard A. Friesner et collaborateurs.
- Approche SP-basis Chemical Potential Equalization (CPE) développée par R. Chelli et P. Procacci.
- Champ de force polarisable CHARMM développé par S. Patel (Université du Delaware) et C.L. Brooks III (Université du Michigan).
- Champ de force polarisable AMBER développé par Jim Caldwell et collaborateurs.
Champs de force polarisables basés sur des multipôles distribués
- SIBFA (Sum of Interactions Between Fragments Ab initio computed)[35]. Champ de force pour petites molécules et protéines flexibles, développé par Nohad Gresh (Université Paris V René Descartes) et Jean-Philip Piquemal (Université Paris VI Pierre et Marie Curie). SIBFA est un procédé de mécanique moléculaire formulé et calibré sur la base de calculs ab initio sur des supermolécules. Son but est de permettre des calculs fiables et simultanés à la fois sur les énergies conformationnelles et intermoléculaires qui induisent les propriétés de liaison spécifiques des molécules d'intérêt biologique et pharmaceutique. Cette procédure permet un traitement précis des métaux de transition. L'inclusion d'une contribution de champ de ligand permet des calculs sur les protéines métalliques « couches ouvertes ».
- AMOEBA (Atomic Multipole Optimized Energetics for Biomolecular Applications). Champ de force développé par Pengyu Ren (Université du Texas à Austin) et Jay W. Ponder (Université de Washington).
- ORIENT procedure. Champ de force développé par Anthony J. Stone (Université de Cambridge) et collaborateurs.
- Non-Empirical Molecular Orbital (NEMO) procédé développé par Gunnar Karlström et collaborateurs à l'Université de Lund (Suède).
Champs de force polarisables basés sur la densité
- Gaussian Electrostatic Model (GEM)[35] - [36] - [37]. Champ de force polarisable développé par Thomas A. Darden et G. Andrés Cisneros au NIEHS et Jean-Philip Piquemal (Université Paris VI Pierre et Marie Curie).
- Procédure de polarisation basée sur l'approche Kim-Gordon développée par Jürg Hutter et collaborateurs (Université de Zurich).
Champs de force polarisables basés sur la théorie de la polarisation de liaison
- COSMOS-NMR. Ce champ de force hybride QM/MM permet des calculs en mécanique quantique explicites des propriétés électrostatiques en utilisant les orbitales de liaison localisées avec le formalisme de la théorie de la polarisation de liaison rapide[38]. La fluctuation de charge atomique est possible pour chaque pas de dynamique moléculaire.
Champs de force réactifs
- ReaxFF. Champ de force réactif développé par Adri van Duin, William Goddard et collaborateurs. Il est rapide, transférable et constitue une méthode numérique intéressante pour les simulations à l'échelle atomique de réactions chimiques.
- EVB (empirical valence bond). Ce champ de force réactif, introduit par Warshel et collaborateurs, est probablement l'une des méthodes les plus fiables et physiquement cohérente d'utilisation des champs de force de modéliser les réactions chimiques des différents environnements. L'EVB facilite les calculs des énergies libres d'activation dans les phases condensées et les enzymes.
- RWFF. Champ de force réactif pour l'eau développé par Detlef W. M. Hofmann, Liudmila N. Kuleshova et Bruno D'Aguanno. Le champ de force rapide, reproduisant les données expérimentales de diffusion de neutron de manière précise, et permet de simuler la formation ou la rupture de liaison de l'eau et des acides[39].
Champs à gros grains
- Virtual atom molecular mechanics (VAMM). Champ de force développé par Korkut et Hendrickson. VAMM est un champ de force à gros grains spécifiquement construit pour des simulations de mécanique moléculaire comme des transitions conformationnelles à large échelle basées sur des interactions d'atomes Cα. C'est un champ de force basé sur des préconnaissances et formulé afin d'identifier les caractéristiques dépendant de la structure secondaire et sur l'information de contact spécifique aux résidus dans les protéines[40].
Autres
- VALBOND. Fonction construite pour la modification d'angle basée sur la théorie de la liaison de valence et marche pour de grandes distorsions d'angles, les molécules hypervalentes et les complexes de métaux de transition. Elle peut être introduite dans d'autres champs de force (comme CHARMM et UFF).
Modélisation de l'eau
L'ensemble des paramètres utilisé pour modéliser l'eau ou les solutions aqueuses (de manière basique un champ de force pour l'eau) est appelé modèle d'eau. L'eau a beaucoup attiré l'attention en raison de ses propriétés inhabituelles et de son rôle majeur comme solvant. De nombreux modèles d'eau ont été proposés, comme : TIP3P, TIP4P, SPC, Flexible SPC ou ST2.
Voir aussi
- chimie numérique
- mécanique moléculaire
- dynamique moléculaire
- modélisation moléculaire
- potentiel statistique
Notes et références
- (en) Ponder JW and Case DA. (2003) Force fields for protein simulations. Adv. Prot. Chem. 66: 27-85.
- (en) Warshel A, Sharma PK, Kato M and Parson WW (2006) Modeling Electrostatic Effects in Proteins. Biochim. Biophys. Acta 1764:1647-1676.
- (en) Schutz CN. and Warshel A. 2001. What are the dielectric "constants" of proteins and how to validate electrostatic models? Proteins 44: 400-417.
- (en) Israelachvili, J.N. 1992. Intermolecular and surface forces. Academic Press, San Diego.
- (en) Leckband, D. and Israelachvili, J. (2001) Intermolecular forces in biology. Quart. Rev. Biophys. 34: 105-267.
- (en) Koehl P. and Levitt M. (1999) A brighter future for protein structure prediction. Nature Struct. Biol. 6: 108-111.
- (en) Brunger AT and Adams PD. (2002) Molecular dynamics applied to X-ray structure refinement. Acc. Chem. Res. 35: 404-412.
- (en) Guntert P. (1998) Structure calculation of biological macromolecules from NMR data. Quart. Rev. Biophys. 31: 145-237.
- (en) Tramontano A. and Morea V. 2003. Assessment of homology-based predictions in CASP5. Proteins. 53: 352-368.
- (en) Gohlke H. and Klebe G. (2002) Approaches to the description and prediction of the binding affinity of small-molecule ligands to macromolecular receptors. Angew. Chem. Internat. Ed. 41: 2644-2676.
- (en) Edgcomb SP. and Murphy KP. (2000) Structural energetics of protein folding and binding. Current Op. Biotechnol. 11: 62-66.
- (en) Lazaridis T. and Karplus (2000) Effective energy functions for protein structure prediction. Curr. Op. Struct. Biol. 10: 139-145
- (en) Levitt M. and Warshel A. (1975) Computer Simulations of Protein Folding, Nature 253: 694-698
- (en) Gordon DB, Marshall SA, and Mayo SL (1999) Energy functions for protein design. Curr. Op. Struct. Biol. 9: 509-513.
- (en) Mendes J., Guerois R, and Serrano L (2002) Energy estimation in protein design. Curr. Op. Struct. Biol. 12: 441-446.
- (en) Rohl CA, Strauss CEM, Misura KMS, and Baker D. (2004) Protein structure prediction using Rosetta. Meth. Enz. 383: 66-93.
- (en) Lomize AL, Pogozheva ID, Lomize MA, Mosberg HI (2006) Positioning of proteins in membranes: A computational approach. Protein Sci. 15, 1318-1333.
- (en) Lomize A.L., Reibarkh M.Y. and Pogozheva I.D. (2002) Interatomic potentials and solvation parameters from protein engineering data for buried residues. Protein Sci., 11:1984-2000.
- (en) Murphy K.P. and Gill S.J. 1991. Solid model compounds and the thermodynamics of protein unfolding. J. Mol. Biol., 222: 699-709.
- (en) Shakhnovich, E.I. and Finkelstein, A.V. (1989) Theory of cooperative transitions in protein molecules. I. Why denaturation of globular proteins is a first-order phase transition. Biopolymers 28: 1667-1680.
- (en) Graziano, G., Catanzano, F., Del Vecchio, P., Giancola, C., and Barone, G. (1996) Thermodynamic stability of globular proteins: a reliable model from small molecule studies. Gazetta Chim. Italiana 126: 559-567.
- (en) Myers J.K. and Pace C.N. (1996) Hydrogen bonding stabilizes globular proteins, Biophys. J. 71: 2033-2039.
- (en) Scholtz J.M., Marqusee S., Baldwin R.L., York E.J., Stewart J.M., Santoro M., and Bolen D.W. (1991) Calorimetric determination of the enthalpy change for the alpha-helix to coil transition of an alanine peptide in water. Proc. Natl. Acad. Sci. USA 88: 2854-2858.
- (en) Gavezotti A. and Filippini G. (1994) Geometry of intermolecular X-H...Y (X,Y=N,O) hydrogen bond and the calibration of empirical hydrogen-bond potentials. J. Phys. Chem. 98: 4831-4837.
- (en) Norman L. Allinger. Conformational analysis. 130. MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms. J. Am. Chem. Soc. 1977, 99, 8127-8134.
- (en) MM2 and MM3's home page
- (en) Allinger, N. L., Yuh, Y. H., & Lii, J-H. (1989) Molecular Mechanics. The MM3 Force Field for Hydrocarbons. 1. J. Am. Chem. Soc. 111, 8551-8565.
- (en) Lii, J-H., & Allinger, N. L. (1989a) Molecular Mechanics. The MM3 Force Field for Hydrocarbons. 2. Vibrational Frequencies and Thermodynamics, J. Am. Chem. Soc. 111, 8566-8575.
- (en) Lii, J-H., & Allinger, N. L. (1989b) Molecular Mechanics. The MM3 Force Field for Hydrocarbons. 3. The van der Waals Potentials and Crystal data for Aliphatic and Aromatic Hydrocarbons, J. Am. Chem. Soc. 111, 8576-8582.
- (en) Schaumann, T., Braun, W. and Wutrich, K. (1990) The program FANTOM for energy refinement of polypeptides and proteins using a Newton-Raphson minimizer in torsion angle space. Biopolymers 29: 679-694.
- (en) Margit Möllhoff, Ulrich Sternberg (2001) Molecular Mechanics with fluctuating atomic charges - a new force field with semi-empirical charge calculation J. Mol. Model. 7: 90-102
- (en) Warshel A (1973). Quantum Mechanical Consistent Force Field (QCFF/PI) Method: Calculations of Energies, Conformations and Vibronic Interactions of Ground and Excited States of Conjugated Molecules, Israel J. Chem. 11: 709.
- (en) Warshel A and Levitt M (1974). QCFF/PI: A Program for the Consistent Force Field Evaluation of Equilibrium Geometries and Vibrational Frequencies of Molecules, QCPE 247, Quantum Chemistry Program Exchange, Indiana University.
- (en) Warshel A. and Levitt M. (1976) Theoretical Studies of Enzymatic Reactions: Dielectric Electrostatic and Steric Stabilization of the Carbonium Ion in the Reaction of Lysozyme, J. Mol. Biol. 103: 227-249
- (en) N. Gresh, G. A. Cisneros, T. A. Darden and J-P Piquemal(2007) Anisotropic, polarizable molecular mechanics studies of inter-, intra-molecular interactions, and ligand-macromolecule complexes. A bottom-up strategy, J. Chem. Theory. Comput. 3: 1960
- (en) J.-P. Piquemal, G. A. Cisneros, P. Reinhardt, N. Gresh and T. A. Darden (2006), Towards a Force Field based on Density Fitting., J. Chem. Phys. 124: 104101
- (en) G. A. Cisneros, J-P. Piquemal and T. A. Darden (2006), Generalization of the Gaussian Electrostatic Model: extension to arbitrary angular momentum, distributed multipoles and speedup with reciprocal space methods, J. Chem. Phys. 125:184101
- (en) Ulrich Sternberg, Frank-Thomas Koch and Margit Möllhoff (1994) New Approach to the Semiempirical Calculation of Atomic Charges for Polypeptides and Large Molecular Systems J. Comp. Chem. 15: 524-531
- (en) D.W.M. Hofmann, L. N. Kuleshova, B. D'Aguanno (2007) A new reactive potential for the Molecular Dynamics simulation of liquid water. Chem.Phys.Lett. 448: 138-143
- (en) Korkut A, Hendrickson WA (2009) A force field for virtual atom molecular mechanics of proteins. Proc Natl Acad Sci USA 106:15667–15672
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Force field (chemistry) » (voir la liste des auteurs).