Spectrométrie de masse à rapport isotopique
La spectrométrie de masse à rapport isotopique (IRMS, pour isotope-ratio mass spectrometry) est une spectrométrie de masse avec laquelle il est possible de mesurer l'abondance relative des différents isotopes d'un même élément chimique dans un échantillon donné. Cette technique est utilisée pour mesurer les rapports n'impliquant que des isotopes non radiogéniques (paléothermométrie et traçage : 2H/1H, 13C/12C, 15N/14N, 18O/16O, etc.) ou impliquant au moins un isotope radiogénique (datation et traçage : 14C/12C, 40Ar/39Ar, 87Sr/86Sr, 206Pb/204Pb, etc.).
Cette technique fut commercialisée dans les années 2000 et ses applications ne cessent de grandir. L'IRMS est utilisée dans plusieurs domaines, tels que les sciences de la Terre, la médecine légale, l'industrie pharmaceutique, la biochimie, la lutte antidopage et les sciences environnementales.
Histoire
L’IRMS découle de l’invention du spectromètre de masse proposé pour la première fois par Joseph John Thomson en 1912. L’IRMS été principalement développé par Alfred Nier (en) dans les années 1930-1940. Cette technique a commencé à être popularisée dans les sciences géologiques peu de temps après la Deuxième Guerre mondiale. En 1939, la diminution fondamentale du rapport 13C/12C dans les échantillons organiques a été reconnue à l’aide de cette technique. Des images de l’instrument original développé par Alfred Nier sont présentes au Musée national d'histoire américaine. Cet instrument était composé d’un analyseur de masse à secteur magnétique à 180° et d’un détecteur multiple. La disposition fondamentale des composantes de l’IRMS n’a étonnamment pas changé depuis 1940. De même, bien que l’IRMS soit connue depuis les années 1940, cette technique est tout de même considérée comme une technique moderne et innovatrice étant donné le développement d’interfaces à débit continu, ce qui facilite le couplage de l’IRMS à d’autres instruments analytiques tels qu’un analyseur élémentaire et un chromatographe en phase gazeuse[1].
Origine de l'abondance des isotopes
Au tout début du premier éon de l'histoire de la Terre (Hadéen), la moyenne du rapport isotopique de chacun des éléments a été fixée. En effet, la variation se produit en fonction d’enrichissement ou d’appauvrissement des isotopes les plus lourds par rapport aux valeurs moyennes. Par exemple, les plantes utilisent le CO2 de l’atmosphère terrestre comme source de carbone et il y a plusieurs facteurs qui influencent l’enrichissement ou l’appauvrissement en carbone 13. Par exemple, les plantes monocotylédones comme le sucre de canne, le maïs, les plantes tropicales, les plantes du désert et les plantes marines utilisent un cycle photosynthétique. Ces plantes ont des valeurs approximatives de δ13C qui varient entre -8 et -20‰ [2]. Les plantes dicotylédones, comme les plantes fleuries, le blé, le riz et utilisent un autre cycle photosynthétique qui donne des valeurs de δ13C qui varient entre -22 et -35‰ [2]. Il y a d’autres facteurs qui peuvent influencer ces facteurs génétiques, comme la température, la pluie, le temps d’exposition au soleil, etc. Ces facteurs influencent la cinétique de diffusion du dioxyde de carbone[2].
En connaissant ces valeurs de rapport isotopique, il est donc possible de trouver la provenance de la nourriture des humains. En effet, les humains ne peuvent ingérer que du carbone par les matières végétales ou animales, donc les rapports isotopiques du carbone dans un animal reflètent les rapports isotopiques des sources de nourriture. Par exemple, les Européens mangent plus de dicotylédones (blé, orge et seigle), causant un rapport isotopique du carbone 13 correspondant entre -22 et -35‰ tandis que les Américains mangent plus de monocotylédones (sucre et maïs), causant un delta qui se situe entre -8 et -20‰[2].
Les rapports isotopiques sont mesurés avec la notation delta (δ), en partie pour mille (‰) de façon relative aux standards universels (CRM) à l’aide de l’équation 1[2].
- δ = 1000 La valeur de est le rapport de l’abondance du plus lourd isotope mineur et du plus léger isotope majeur (par exemple 13C/12C). Pour la valeur de , elle est établie selon les standards, parce qu’elle représente un matériel stable hautement enrichi en isotope plus lourd (mineur). En effet, la plupart des éléments analysés sont appauvris en isotope plus lourd, comme le carbone 13, comparativement aux standards où le carbone 13 est enrichi. Il sera donc normal d’obtenir une valeur de delta négative, car sera plus grand que . Un exemple d’application de la valeur de delta est dans les contrôles antidopage. Une grande valeur de delta d’un stéroïde révèle un taux élevé d’isotope, comportement retrouvé dans les molécules synthétiques[2].



Selon le type d'analyse, plusieurs méthodes de séparations peuvent être employées. Les méthodes sont choisies en fonction du type d'analyte à analyser et de la matrice. Pour ce faire, il faut choisir la bonne méthode d'étalonnage pour que l'analyse soit faite de façon optimale. Par exemple, il est possible de prioriser un étalonnage interne pour veiller au bon fonctionnement de l'appareil. Donc, un étalon interne est ajouté à chaque analyse afin d'éliminer tout biais ou erreur systématique dans les mesures. De plus, des matériaux de référence certifiés (CRM) sont utilisés pour calibrer les appareils. Des standards sont reconnus internationalement comme Vienna Pee Dee Belemnite (VPDB) pour le carbone, Vienna Canyon Diablo Troilite météorite (V-CDT) pour le soufre, l'Eau océanique moyenne normalisée de Vienne ou Vienna Standard Mean Ocean Water (VSMOW) pour l’oxygène et l’hydrogène et l’air ambiant pour l’azote[2]. Chaque méthode d'analyse doit être validée selon plusieurs critères. Pour plus d'information, voir la section Validation de la méthode ci-dessous.
Fonctionnement
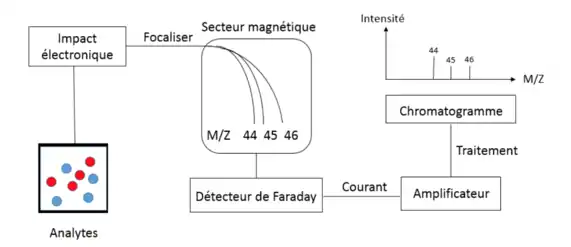
L’IRMS est très semblable à une séparation couplée avec un spectromètre de masse. Les analytes peuvent être séparés soit par chromatographie liquide ou gazeuse, suivie d’une analyse élémentaire. Ils sont ensuite oxydés ou brûlés, les convertissant en gaz simple, par exemple le CO2, N2, H2, SO2. Ces gaz sont ensuite analysés par un spectromètre de masse pour déterminer l’abondance des isotopes. Pour le dioxyde de carbone, il y aura des moniteurs dans le spectromètre de masse à des rapports masse sur charge (m/z) de 44, 45, 46, qui correspondent aux combinaisons isotopiques des ions produits par le CO2. Il sera donc possible de déterminer le rapport isotopique des isotopes 12C, 13C, 16O, 17O et 18O dans différentes combinaisons et proportions. Les signaux sont ensuite traités par un ordinateur pour afficher les différents rapports analysés.
Composantes
Les composantes de cet instrument dépendent de la méthode de séparation utilisée. En général, ce type de système nécessite une chambre de combustion, une interface et un spectromètre de masse. La chambre de combustion consiste en une atmosphère remplie d’oxygène et un réacteur en quartz. La température de combustion peut atteindre un sommet de 1 800 °C. Un deuxième réacteur en quartz, chauffant à 650 °C, permet d’éliminer l’excès d’oxygène et de réduire les NOx et N2. L’eau formée lors de la combustion est séparée via une trappe à eau constituée par exemple de perchlorate de magnésium (en). Les ions sont ensuite envoyés dans le spectromètre de masse pour l'analyse[3].
Couplage
Le spectromètre de masse à rapport isotopique à la capacité de se coupler avec différents types de séparation. En fonction de l'analyte à étudier, une méthode d'analyse peut être favorisée. La prochaine section démontre les différentes façons de coupler l’IRMS avec les méthodes de séparations, entre autres l'analyse élémentaire, la chromatographie gazeuse et liquide.
EA-IRMS
L’analyse élémentaire permet d’analyser l’échantillon avec des données assez représentatives pour tout le contenu de l’analyte. Sans préparation adéquate, cette méthode ne divulgue pas comment tous les constituants contribuent aux données obtenues. Il existe deux types d’analyses élémentaires particulièrement utilisées en EA-IRMS. Il s’agit de l’analyse pour les isotopes du carbone et l’azote connu sous le nom de EA-IRMS, ainsi que les analyses pour les isotopes de l'hydrogène et l’oxygène connues sous plusieurs noms différents : Thermal conversion EA-IRMS (TC/EA-IRMS), high-température conversion IRMS (HTC-IRMS), high-temperature pyrolysis IRMS (HTP-IRMS) et high-temperature carbon reduction IRMS (HTCRIRMS)[4].
Ces deux analyses peuvent être divisées en quatre étapes. La première est la combustion, ou conversion thermale, de l’échantillon, à l’aide d’un analyseur élémentaire, suivie de l’introduction des gaz résultants dans la source d’ions du spectromètre de masse. Troisièmement, le gaz est ionisé par la source d'ions, donc la séparation et la détection des ions dans le spectromètre de masse permettent d’acquérir les données nécessaires à l’analyse EA-IRMS. La quatrième étape consiste à traiter les données brutes provenant du spectromètre de masse pour obtenir des valeurs de rapports isotopiques. Ces analyses peuvent être effectuées sur des échantillons solides et sur des liquides non volatils à l’aide d’un analyseur élémentaire utilisant des capsules d’argent ou d’étain, tandis que les liquides avec de faibles viscosités peuvent être injectés en utilisant un système d’injection[4].
Pour l’analyse du carbone et de l’azote, l’échantillon est premièrement passé dans un réacteur de combustion, puis dans un réacteur de réduction, suivi par un système pour éliminer l’eau des gaz produits et parfois par une colonne de chromatographie gazeuse pour séparer le CO2 et le N2 résultant. La combustion se produit dans un réacteur en quartz sous atmosphère de O2 pour transformer les molécules organiques en CO2, en NOx et en H2O. Ce réacteur contient habituellement aussi du Cr2O3 et du Cr3O4 + Ag pour lier les souffres et les halogènes, mais plusieurs variations de réactifs peuvent être utilisés pour des applications spécifiques. Le réacteur à combustion est généralement à des températures entre 900 et 1 050 °C, mais la combustion des capsules d’étain peut augmenter la température de la capsule jusqu’à 1 800 °C. Étant donné la formation de cendres et de résidus résultants de la combustion de l’échantillon et de la capsule d’étain, il est recommandé d’utiliser un creuset en quartz que l’on remplace chaque 50 à 150 échantillons pour éviter de salir l’appareil. Le réacteur de réduction en quartz à 650 °C contient généralement du cuivre de haute pureté pour réduire les NOx, l’oxygène en excès et le N2, mais des variations de températures et de réducteur peuvent être faites pour des applications spécifiques[1].
Pour l’analyse de l’hydrogène et de l’oxygène, le réacteur de combustion est à une température entre 1 350 °C et 1 450 °C pour transformer les composés organiques et inorganiques en H2, N2 et CO. Les gaz sont ensuite séparés par chromatographie gazeuse puisque N2 est isobarique avec CO (m/z 28) et est connu pour influencer l’ionisation de H2[1]. Lorsque les échantillons ont besoin d’être séparés préalablement, il est plus efficace d’utiliser la chromatographie gazeuse (GC) pour les échantillons volatils. Les produits provenant de la colonne GC sont transportés vers une chambre de combustion. Par contre, il est aussi possible d’envoyer des composés vers un spectromètre de masse externe ou un détecteur à ionisation de flamme. Ensuite, cette chambre est majoritairement constituée d’un tube d’aluminium contenant du cuivre, du nickel et du platine. Les composés sont brûlés à très haute température et produisent une combinaison de CO2, NOx et H2O. Comme en analyse élémentaire, l’échantillon est transporté dans une chambre de réduction où les oxydes nitreux sont convertis en N2 et l’excès de O2 est retiré. De plus, l’eau en retiré à l’aide d’une trappe chimique et d’hélium. Par la suite, l’échantillon est introduit dans la source d’ions de l’IRMS à un débit très stable d’approximativement 0,5 mL/min.
LC-IRMS
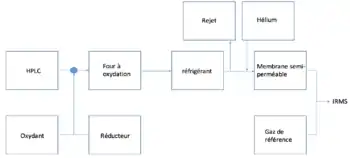
La chromatographie liquide (LC) couplée à de la spectrométrie de masse à rapport isotopique est typiquement destinée à l’analyse du carbone. Lorsque la solution quitte la colonne, elle est dirigée vers une interface d’oxydation destinée aux produits chimiques humides[2]. En effet cette méthode convertit les produits organiques en produit gazeux du CO2. Par contre, il faut prendre des précautions supplémentaires pour réduire les interférences. Par exemple, il faut s’assurer que la phase mobile ne contient pas de composés oxydables qui pourraient influencer les résultats. Pour ce faire, l’éluant est mélangé à un agent oxydant comme du persulfate d'ammonium et un catalyseur comme de l’acide phosphorique avec du nitrate d’argent[2]. Le tout est alors oxydé dans un réacteur à capillaire où les produits organiques sont convertis en CO2. Une membrane échangeuse séparera les différents gaz (vapeur d’eau, oxygène, argon, etc.) et le tout sera transféré au travers d’une membrane perméable aux gaz à l’aide d’hélium à contre-courant. Le CO2 contenu dans l’hélium est alors séché et est transmis à l’appareil via un open split. La détermination du rapport isotopique 13C/12C d’un échantillon peut donc être faite avec une méthode de très haute précision.
GC-IRMS
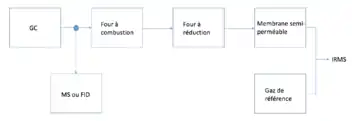
Lorsque les échantillons ont besoin d’être séparés préalablement, il est plus efficace d’utiliser la chromatographie gazeuse (GC) pour les échantillons volatils. Les produits provenant de la colonne GC sont transportés vers une chambre de combustion. Par contre, il est aussi possible d’envoyer des composés vers un spectromètre de masse externe ou un détecteur à ionisation de flamme. Ensuite, cette chambre est majoritairement constituée d’un tube d’aluminium contenant du cuivre, du nickel et du platine. Les composés sont brûlés à très haute température et produisent une combinaison de CO2, NOx et H2O. Comme en analyse élémentaire, l’échantillon est transporté dans une chambre de réduction où les protoxyde d'azoteoxydes nitreux sont convertis en N2 et l’excès de O2 est retiré. De plus, l’eau est retirée à l’aide d’une trappe chimique et d’hélium. Par la suite, l’échantillon est introduit dans la source d’ions de l’IRMS à un débit très stable d’approximativement 0,5 mL/min.
L'IRMS vs MS et ICP-MS
Les spectromètres de masse communs utilisant des quadripôles (Q), pièges à ions quadripolaires (QIT) ou des temps de vols (TOF), ne produisent pas la sensibilité et la précision requise pour détecter les différences subtiles de l’abondance naturelle des isotopes.
Par contre ces instruments peuvent être utilisés avec des échantillons ayant subi une dilution isotopique, une technique dans lesquelles les isotopes plus lourds sont délibérément enrichis bien au-delà de leurs niveaux naturels par l'analyste. La mesure des abondances isotopiques naturelles nécessite un instrument spécialisé comme un spectromètre de masse à secteur magnétique multicollecteur, tel un IRMS. En effet, des tests sur la précision et l’exactitude montrent que l’appareil obtient une précision de 0,1‰ avec la plus petite limite de détection contenue entre 0,07 et 0,35 µg/L. Les principales sources d’erreurs de reproductibilité sont la stabilité du courant ionique, le temps mort et les fluctuations dans les composantes électroniques[2].
Une autre technique pouvant être utilisé pour déterminer le rapport isotopique est celle de la spectroscopie de masse à plasma à couplage inductif à collecteur multiple, ou communément multiple collector inductively coupled plasma mass spectrometry (MC-ICP-MS). En fait, l’étude de Clough et al.[5] a démontré qu’il s’agit d’une technique a haute performance pour mesurer le δ34S des échantillons liquides et solides en utilisant Si comme étalon interne pour la correction des effets de polarisation de masse. Cette technique est limitée par les instabilités du plasma et la performance de l'acquisition des données en mode séquentiel, ce qui engendre de grandes variations dans les abondances isotopiques.
Détecteur
Dans cette technique d’analyse, l'analyseur est de type spectromètre de masse. Les analytes sont d’abord ionisés par impact électronique. Des électrons de haute énergie, soit de 70 eV, collisionnent avec les différents gaz. L'ionisation par nébulisation n'est pas assez énergétique pour ioniser les gaz, car ces molécules stables possèdent des liens forts. Cet impact les ionise selon la réaction suivante : A(g)+ e− → A+(g)+ 2e− , où A représente le gaz analysé. Les ions quittent la source et ils sont focalisés et accélérés sous une différence de potentiel. Ils passent ensuite dans un champ magnétique (secteur magnétique), séparant ainsi les différents ions selon leur masse et leur charge, avant de frapper les détecteurs de Faraday. Ce type de détecteur est formé d'un métal conducteur. Lorsqu’un ion frappe ce dernier, un courant est produit. Le courant induit dépend également du nombre d'ions interceptés. Le courant généré par ses détecteurs est ensuite amplifié et analysé en fonction du temps par le système de données IRMS. Ce dernier trace un chromatogramme pour les ions spécifiques d’un rapport m/z. L’aire sous la courbe est proportionnelle au nombre d’électrons détecté. Comme mentionné auparavant, un analyseur de masse de type quadripôle, temps de vol ou piège à ions quadripolaire ne possèdent pas la précision et la sensibilité pour détecter les différences subtiles entre les abondances des isotopes[4].
Application
L'IRMS est appliqué dans plusieurs domaines en raison de sa capacité à analyser des matériaux. Les principales applications de l’IRMS sont dans le domaine légal, pharmaceutique, alimentaire, environnemental, ainsi que dans les dépistages dans le milieu sportif et dans la recherche biochimique sur l’alimentation et le métabolisme de certains animaux.
Domaine légal
Dans le domaine légal, l’IRMS peut être utilisé pour confirmer ou infirmer la source d’un échantillon de terre. Par exemple, cette technique est utile pour disculper un suspect sur lequel l’échantillon a été recueilli, pour identifier la source géographique d’un produit d’exportation et pour vérifier si un composé (par exemple l'Acide gamma-hydroxybutyrique (GHB)) est présent dans le sang, l’urine ou les cheveux d’un individu provient de source endogène ou exogène[2].
De plus, cette technique peut être utilisée pour déterminer si un échantillon de drogue saisi vient de la même source que d’autres échantillons ou s’il provient d’une nouvelle source de production jusqu’ici inconnue. L’IRMS peut aussi aider à déterminer la provenance géographique du corps d’une victime d’une catastrophe ou d’une explosion s’il n’est pas possible de prélever un échantillon d’ADN intact. En effet, en mesurant le rapport isotopique d’un échantillon et en comparant ce rapport isotopique avec un échantillon similaire provenant de la location géographique présumée de la source, il est possible de confirmer ou d’infirmer si l’échantillon provient de cette région du monde[2].
Domaine pharmaceutique
Dans les domaines pharmaceutiques et alimentaires, l’IRMS est utilisée pour déterminer si un produit concorde avec les informations étiquetées. Il est par exemple possible de savoir si les sources de sucres dans un produit sont celles affichées, si ce sont des sucres artificiels ou s'ils proviennent d’autres végétaux que ceux attendus. Cette technique pour analyser les sucres a été développée à l’aide d’un LC-MS. Par exemple, l'importation de certains aliments venant de pays spécifiques a été bannie auparavant. L'IRMS permet donc de déterminer le lieu de provenance de ces aliments, confirmant ou non l'importation légale. Une autre application peut être de déterminer si le CO2 présent dans une boisson gazeuse provient d’une fermentation ou s’il provient d’une source externe telle qu’un cylindre de CO2, ce qui permet au producteur du produit de diminuer et de simplifier la production de son produit[2].
Domaine des sciences de la Terre
Le domaine environnemental utilise cette technique entre autres pour associer un contaminant dans une région à la production d’une usine en particulier. Les archéologues peuvent également l'utiliser pour déterminer la provenance d'une roche sédimentaire ou pour identifier la source d'un échantillon d'eau. En analysant des échantillons de terre polluée et de terre près d’une usine par GC-IRMS, des chercheurs ont prouvé que les contaminants du sol ne provenaient pas de l’usine, mais plutôt d’une source naturelle de charbon[2].
Domaine sportif
Dans le milieu sportif, l’IRMS est très utilisée pour déterminer si un stéroïde dopant présent dans le sang, l’urine ou les cheveux d’un sportif provient d’une source endogène ou exogène. Des niveaux élevés de certains stéroïdes peuvent parfois être détectés chez un athlète sans qu’il ait fait usage de produits dopants, ce qui peut indiquer une anomalie génétique, ou une alimentation augmentant la production naturelle de stéroïdes. Étant donné la différence de la provenance de carbone entre la synthèse biochimique d’un stéroïde dans le corps humain et une synthèse chimique, les rapports isotopiques dans des stéroïdes endogènes et exogènes diffèrent. En fonction de l’abondance des isotopes d’une molécule organique, par exemple la testostérone, les centres antidopage peuvent confirmer ou non la présence de cette molécule provenant d'une source synthétique ou naturelle. De ce fait, une molécule endogène possède un plus petit rapport isotopique qu’une molécule synthétique. Après un certain rapport isotopique considéré légal, il est possible de confirmer la prise endogène ou exogène d'un stéroïde anabolisant[2].
Domaine archéologique
Dans la recherche en biochimie, en nutrition et sur le métabolisme, L’IRMS a été utilisée pour obtenir des preuves que les Incas avaient une alimentation basée principalement basée sur le maïs. Pour ce faire, les chercheurs ont analysé les rapports isotopiques du carbone et de l’azote dans le collagène présent dans des os prélevés sur des sites archéologiques. Pour compléter ces résultats, ils ont aussi analysé ces rapports isotopiques du carbone et de l’azote dans des muscles et de la peau prélevée sur des momies datant des années 1490-1640 et provenant de la vallée Ayacucho au Pérou. Dans des cas comme celui-ci où l’on travaille avec des acides aminés ou des acides gras non volatils, l’échantillon doit passer par plusieurs étapes de préparation et de dérivation avant d’être introduit dans le GC-IRMS[2].
Domaine de la zoologie
En zoologie, l’IRMS a été utilisée pour étudier la chronologie des habitudes alimentaires et migratoires des éléphants en analysant les rapports d’isotopes du carbone et de l’azote présents dans leurs poils. Les rapports isotopiques moyens obtenus ont montré des différences d’une saison à l’autre. De plus, les chercheurs ont remarqué qu’il était possible de déterminer si un éléphant avait été impliqué dans les dégâts d’une culture. Si la culture est dans une autre classe métabolique que les espèces natives, les rapports isotopiques du carbone ingéré par un éléphant ayant mangé une récolte seront différents que les rapports isotopiques du carbone ingéré par un éléphant ayant consommé des plantes natives. En analysant les plumes d’espèces d’oiseau, il a aussi été possible de développer une relation entre leurs migrations et les rapports isotopiques de carbone et d’hydrogène[2].
Avantage et inconvénient
Cette technique d'analyse offre un avantage supérieur lorsqu'il s'agit à comparer des molécules avec des propriétés physique et chimique semblables. Les différences d'isotope révèlent des informations sur son origine ainsi que sur son parcours. Sur le plan judiciaire, cette méthode d'analyse peut innocenter un suspect ou non. Sa grande précision et reproductibilité est alors un aspect important. De plus, sa grande versatilité à être associé à divers systèmes de séparation lui permet d'analyser pratiquement tous les échantillons, soit dans une phase liquide ou gazeuse.
L'inconvénient de cette technique est la complexité du matériel nécessaire afin d'effectuer l'analyse. Comme mentionné, l'appareil doit être doté d'une haute précision. Or, ce matériel est très coûteux. L'entretien de l'appareil est primordial pour conserver sa précision et son exactitude. De plus, cette technique d'analyse est spécifique lorsqu'il s'agit d'observer les rapports isotopiques, car il n'y a pas d'autres techniques qui peuvent donner des résultats aussi précis.
Validation de la méthode
Chaque méthode d'analyse doit être validée selon plusieurs critères afin d'obtenir la précsion qu'elle nécessite. Le laboratoire à la responsabilité de préparer ce plan de validation. Ce plan dépend de la nature de l'échantillon à analyser ainsi que le matériel et les paramètres employés. Le plan de validation doit inclure plusieurs paramètres, entre autres la linéarité, la stabilité, la reproductibilité de l'appareil, la sélectivité et la robustesse. La linéarité est déterminée en utilisant des échantillons réels avec addition d'un gaz connu, soit une méthode d'étalonnage interne. L'expérience doit être reproductible dans différents jours, par différents expérimentateurs, laboratoires et systèmes. De plus, l'appareil doit démontrer une reproductibilité entre chaque mesure du même échantillon. La robustesse démontre les effets de variation des paramètres de l'instrument, par exemple la température ou le débit d'approvisionnement[4]. Pour vérifier la reproductibilité de l'appareil, il est possible d'utiliser des alcanes à différentes concentrations. Si l'appareil est en bonne condition, toutes les concentrations devraient être au même rapport isotopique, car ce rapport est indépendant de la concentration. De plus, à chaque changement de bonbonne de gaz, il faut refaire une validation de la méthode.
Entretien
Une routine de maintenance doit être faite régulièrement pour assurer un roulement continu et des temps d'arrêt minimaux de l'appareil. Une routine typique consiste à nettoyer les plateaux d'échantillonnage, retirer les cendres, remplacer les réacteurs de quartz, replacer les trappes à eau et nettoyer la source d'ion pour s’assurer d’une bonne fluidité[3].
Aussi, il est important de changer la source aux six mois pour une utilisation quotidienne. Pour mener au bon fonctionnement de l’appareil, la maintenance de la pompe mécanique primaire et de la pompe turbo moléculaire est essentielle pour maintenir un vide constant et optimal dans le spectromètre de masse. Une inspection visuelle est également importante pour prévenir des bris de matériel potentiel. Une lacune dans l’entretien peut entraîner un bris et peut provoquer une déficience dans l’exactitude et la précision de l’appareil.
Notes et références
- (en) « Isotope ratio mass spectrometry – history and terminology in brief », Drug testing and analysis, .
- « Isotope ratio mass spectrometry », The Royal Society of Chemistry,
- (en) « Stable isotope ratio mass spectrometry in climate change research », International Journal of Mass Spectromerty, .
- « Good practice guide of isotope ratio mass spectrometry », FIRMS, .
- (en) « δ34S Measurements of Sulfur by Multicollector Inductively Coupled Plasma Mass Spectrometry », Anal. Chem., .