Règle des phases
En physique, et particulièrement en thermodynamique chimique, la règle des phases (ou règle des phases de Gibbs, ou règle de Gibbs) donne le nombre maximum de paramètres intensifs qu'un opérateur peut fixer librement sans rompre l'équilibre d'un système thermodynamique. Ce nombre de paramètres indépendants est appelé variance. Les paramètres sont choisis le plus souvent parmi la pression, la température et les concentrations des diverses espèces chimiques dans les diverses phases présentes.
On doit la règle des phases au physicien américain Willard Gibbs qui l'établit entre 1875 et 1878. Elle fut complétée, en ce qui concerne les systèmes sièges d'équilibres chimiques, par le physicien et chimiste français Pierre Duhem, qui donna le nom de variance au nombre de degrés de liberté déterminé par cette règle[1].
Énoncé de la règle
Dans le domaine de stabilité d’une phase (solide, liquide, gaz) d'un corps pur, un opérateur peut faire varier la température et la pression indépendamment l'une de l'autre et la phase reste stable. Lors d'un changement de phase, où deux phases sont en équilibre, un opérateur peut choisir la température ou la pression et l’autre paramètre est subi : l'opérateur ne peut pas modifier la pression indépendamment de la température, sous peine de se retrouver dans un domaine où seule l'une des phases est présente, donc hors équilibre. Enfin, le point triple d'un corps pur ne laisse aucun degré de liberté car il n'existe qu'à une seule température et une seule pression : l'opérateur ne peut modifier aucun de ces deux paramètres.
La règle des phases de Gibbs généralise cette observation à tout système thermodynamique contenant plusieurs espèces chimiques, siège de plusieurs phases et de plusieurs réactions chimiques. L'équilibre thermodynamique de ce système inclut :
- l'équilibre mécanique, soit l'homogénéité de la pression dans le système : la pression est identique en tout point du système, dans toutes les phases ;
- l'équilibre thermique, soit l'homogénéité de la température dans le système : la température est identique en tout point du système, dans toutes les phases ;
- l'équilibre des phases, soit pour chaque espèce chimique l'homogénéité de son potentiel chimique dans le système : le potentiel chimique de chaque espèce est identique en tout point du système, dans toutes les phases où cette espèce est présente ;
- l'équilibre des réactions chimiques, soit pour chaque équilibre chimique une affinité chimique nulle.
La règle des phases énonce que la variance de ce système à l'équilibre vaut :
| Règle des phases : |

est la variance, ou nombre de degrés de liberté, nombre entier positif ou nul.

- La variance représente le nombre de paramètres intensifs (conditions opératoires) que l'opérateur peut fixer indépendamment les uns des autres. Ces paramètres sont généralement choisis parmi la pression, la température, les fractions molaires ou les concentrations des espèces chimiques dans chacune des phases.
- Si l'opérateur impose plus de paramètres que ce que permet le système, certains de ces paramètres ne sont pas indépendants des autres : le système est surdéterminé. Si l'opérateur peut imposer un nombre de paramètres égal à . Lorsque le système est entièrement déterminé. Lorsque l'opérateur a fixé un nombre de paramètres égal à la variance, tous les autres paramètres se déduisent des équations régissant l'équilibre thermodynamique du système.



est le nombre d'espèces chimiques présentes.

- Les espèces chimiques sont comptées indépendamment de leur état : par exemple l'eau H2O liquide, solide ou gazeuse est considérée comme une seule espèce. Par contre les ions sont comptés comme des espèces indépendantes du sel dont ils sont issus (par exemple NaCl, Na+ et Cl− comptent pour trois espèces distinctes, voir plus bas les exemples de solubilités de sels au paragraphe Exemples).
- D'autre part, même s'ils n'apparaissent pas dans les équations bilans des réactions chimiques, les solvants et les inertes doivent être comptés, puisque leurs concentrations modifient celles des autres espèces chimiques (voir plus bas l'eau dans les exemples de solubilités de sels au paragraphe Exemples). En revanche les catalyseurs et les inhibiteurs, qui n'interviennent ni dans les équilibres de phases, ni dans les équilibres chimiques (ils permettent d'accélérer ou de ralentir les réactions, mais ils ne déplacent pas les équilibres), ne doivent pas être comptés.
est le nombre d'équilibres chimiques indépendants.

- Des équilibres chimiques indépendants signifie qu'aucun des équilibres considérés n'est une combinaison des autres équilibres (voir plus bas l'exemple de la combustion du soufre en présence d'oxygène au paragraphe Exemples).
est le nombre de paramètres, ou conditions opératoires, imposés par l'opérateur.

- L'opérateur fixe généralement un ou plusieurs paramètres parmi la pression, la température et les concentrations des espèces chimiques dans les différentes phases.
- Si à l'issue du calcul , l'opérateur a fixé trop de paramètres, certains d'entre eux ne sont pas indépendants des autres (par exemple, dans l'équilibre liquide-vapeur d'un corps pur l'opérateur ne peut pas fixer la pression indépendamment de la température). Si alors l'opérateur peut encore fixer d'autres paramètres. Si l'opérateur a fixé le nombre de paramètres exact permettant de déterminer entièrement le système (voir plus bas l'exemple des équilibres de phases de l'eau au paragraphe Exemples).
- D'autre part, il peut exister à l'équilibre des contraintes autres que les équilibres mécanique, thermique, chimique et équilibres des phases listés plus haut. Par exemple l'électroneutralité d'une phase liquide contenant des ions impose l'égalité des nombres de charges positives et négatives en solution (voir plus bas les exemples de solubilités de sels au paragraphe Exemples). De même, si les réactifs sont introduits à la stœchiométrie les produits seront eux aussi formés selon la stœchiométrie (voir plus bas l'exemple de la décomposition de l'ammoniac au paragraphe Exemples). On déduira de ces contraintes supplémentaires par l'intermédiaire du nombre .
- Lorsque , le paramètre est appelé variance réduite.

représente l'équilibre mécanique et l'équilibre thermique dont dépend le système.

- En toute rigueur , la pression et la température ont toutes deux une influence sur le système, quel qu'il soit. Mais il arrive que l'un des deux paramètres ne soit pas un facteur d'équilibre sensible. Par exemple, la pression n'a quasiment aucune influence sur les phases condensées (liquide et solide) dans des conditions opératoires proches de la pression atmosphérique : on comptera alors , pour la seule température, en l'absence d'une phase gazeuse. À contrario, l'influence de la pression devient prépondérante par rapport à celle de la température en géophysique.


est le nombre de phases en équilibre dans le système.

- Cette formule est toujours valable si certaines espèces ne sont pas présentes dans toutes les phases considérées (par exemple dans une réaction de précipitation les ions et le solvant ne sont présents qu'en phase liquide) et si certaines phases ne sont constituées que d'un seul corps (le précipité est le seul constituant de la phase solide).
- Les différentes formes cristallines ou moléculaires d'une substance chimique (voir par exemple les diverses formes de la glace d'eau au paragraphe Exemples) comptent pour autant de phases indépendantes, ainsi que, pour les mélanges, les corps solides non miscibles (voir plus bas les exemples de dissolutions simultanées de sels au paragraphe Exemples). Il en est de même pour des liquides non miscibles (par exemple un mélange d'huile et d'eau). Par contre, tous les gaz étant miscibles par diffusion spontanée, il ne peut y avoir au plus qu'une seule phase gaz.
Dans le cas d'un mélange sans contrainte extérieure (), dans lequel aucune réaction chimique n'a lieu () et pour lequel les facteurs d'équilibre sont la pression et la température () on obtient la relation :



Cette relation est utilisée pour calculer la variance dans le cas des équilibres de phases en l'absence de réaction chimique. C'est la forme originelle de la règle, telle qu'établie par Gibbs. Un corps pur, soit , ne peut être en équilibre sous plus de trois phases simultanément, en effet donnerait une variance négative. Par contre, des mélanges de plusieurs espèces chimiques peuvent présenter bien plus de phases liquides[2] et solides en équilibre. En théorie, un mélange de espèces chimiques peut présenter jusqu'à phases en équilibre.



Note - Déplacement et rupture d'équilibre[3].
- Si l'on modifie les conditions opératoires qui ont permis d'établir un équilibre, il y a déplacement d'équilibre lorsque les phases et les espèces chimiques en présence sont les mêmes avant et après la modification. Une rupture d'équilibre a lieu lorsqu'une phase ou une espèce chimique apparait ou disparait à la suite de cette modification.
- Par exemple, il y a déplacement d'équilibre si l'on augmente la température d'un équilibre liquide-vapeur et que les deux phases subsistent ; il y a rupture d'équilibre si le liquide est totalement vaporisé. De même, il y a déplacement d'équilibre d'une solution aqueuse si l'on augmente la concentration en sel et que celui-ci reste totalement dissout ; il y a rupture si un précipité apparait : il constitue à la fois une nouvelle espèce chimique et une nouvelle phase.
- Après déplacement ou rupture d'équilibre le système thermodynamique atteint un nouvel équilibre. Dans le cas d'un déplacement l'état physico-chimique du système thermodynamique est inchangé, seules les pression, température et concentrations ont évolué : la variance ne change pas. À la suite d'une rupture d'équilibre, le système thermodynamique atteint un nouvel état physico-chimique, différent de l'état initial : la variance peut être modifiée ; elle peut ne pas l'être, par exemple lors de la vaporisation totale d'un liquide le nombre de phases est inchangé, mais ce ne sont pas les mêmes avant et après l'opération.
Démonstration de la formule
Le système thermodynamique est décrit par un système de équations et de inconnues. Par définition, la variance vaut .



est le nombre de paramètres intensifs permettant de décrire le système thermodynamique étudié. Il inclut la pression, la température et les potentiels chimiques. On a :
- la pression et la température : en règle générale (il existe des cas où la pression n'est pas un facteur, auxquels cas ) ;
- le nombre total de potentiels chimiques s'il y a espèces chimiques et phases (chaque espèce chimique possède un potentiel chimique dans chacune des phases).

En conséquence :

inclut le nombre de relations entre paramètres intensifs et le nombre de paramètres fixés par un opérateur :
- le nombre de phases correspond au nombre de relations de Gibbs-Duhem : pour chaque phase une relation de Gibbs-Duhem lie entre elles les variations de la pression, de la température et des potentiels chimiques des espèces chimiques dans cette phase ;
- est le nombre d'équilibres chimiques indépendants : chaque équilibre chimique induit que l'affinité chimique correspondante, qui lie entre eux les potentiels chimiques des réactifs et des produits à pression et température données, est nulle ;
- est le nombre de paramètres fixés par un opérateur (pression, température ou concentration, soit les conditions opératoires imposées par l'opérateur) ;
- correspond au nombre de relations que les équilibres de phases induisent entre les potentiels chimiques : pour phases toute espèce chimique possède potentiels chimiques (un dans chacune de ces phases) et à l'équilibre des phases ces potentiels chimiques sont égaux, ce qui induit égalités pour chacune des espèces.


En conséquence :

Ainsi, on calcule :

La démonstration ci-dessus est effectuée en considérant d'emblée les homogénéités de la pression et de la température dans l'ensemble du système à l'équilibre. Il n'a été compté comme paramètres qu'une seule pression et une seule température pour l'ensemble du système. En toute rigueur, il faudrait compter dans une pression et une température par phase, soit un total de pressions et températures, et inclure dans les équilibres mécanique et thermique, soit un total de relations ( égalités de pressions et autant de températures).


Si l'un des composants n'est pas présent dans toutes les phases, il ne possède pas potentiels chimiques. On suppose qu'il n'est présent que dans phases : il possède potentiels chimiques qui induisent égalités à l'équilibre des phases. Les relations de Gibbs-Duhem des phases dans lesquelles le constituant est absent existent toujours mais ne tiennent pas compte de ce constituant (à fortiori la relation de Gibbs-Duhem existe toujours si une phase est constituée d'un corps pur). On a ainsi : et . En calculant on retrouve l'expression de établie précédemment. L'expression de la variance n'est par conséquent pas modifiée, ceci étant généralisable si plusieurs constituants sont absents d'une ou plusieurs phases.






Dans la pratique, un opérateur ne fixe pas le potentiel chimique d'une espèce dans une phase, mais sa concentration ou sa fraction molaire. Dans la démonstration ci-dessus les relations de Gibbs-Duhem peuvent être remplacées par les contraintes imposant que la somme des fractions molaires des espèces chimiques dans une phase est égale à un, la nullité des affinités chimiques peut être remplacée par les lois d'action de masse des équilibres chimiques, l'égalité des potentiels chimiques d'une même espèce dans les différentes phases peut être remplacée, pour les équilibres liquide-gaz idéaux, par la loi de Raoult ou la loi de Henry.
Exemples d'application
Phases d'un corps pur

Les courbes représentent les équilibres de deux phases : si la température est fixée, la pression est subie, et réciproquement. Dans les domaines délimités par les courbes une seule phase est stable : on peut y faire varier la pression indépendamment de la température. Le point critique et le point triple ne peuvent être modifiés.
L'eau peut exister dans trois états : solide, liquide, gaz, auxquels on peut adjoindre le fluide supercritique. On a une seule espèce chimique, l'eau, soit . Le système eau seule n'est le siège d'aucune réaction chimique, soit . Si l'opérateur n'impose aucune condition opératoire alors .
Pour l'eau sous forme de solide seul, de liquide seul, de gaz seul ou de fluide supercritique seul, il n'y a qu'une seule phase, soit :


Il y a donc deux degrés de liberté : à pression donnée la phase étudiée existe seule sur une plage de température plus ou moins étendue. De même, à température donnée la phase étudiée existe seule sur une plage de pression plus ou moins étendue. L'opérateur peut donc imposer la température et la pression indépendamment l'une de l'autre dans le domaine d'existence de la phase étudiée. Si l'opérateur fixe la pression et la température alors et : le système est entièrement déterminé.

Lors des changements d'état on trouve deux phases en équilibre (solide et liquide, ou liquide et gaz, ou solide et gaz), soit :


Il n'y a qu'un seul degré de liberté : à température donnée il n'y a qu'une seule pression d'équilibre, et réciproquement. L'opérateur ne peut donc imposer qu'un seul des deux paramètres pour étudier l'équilibre voulu : la température ou la pression, l'autre paramètre est subi. Si l'opérateur fixe la pression ou la température alors et : le système est entièrement déterminé. Si l'opérateur fixe la pression et la température alors et : l'opérateur impose trop de paramètres.


Au point critique il existe toujours une phase gaz et une phase liquide (), mais elles ne se distinguent plus, elles ont les mêmes propriétés physiques, notamment la même masse volumique. Ceci constitue une contrainte supplémentaire () par rapport à l'équilibre liquide-vapeur existant à des températures et pressions plus basses : la variance réduite est de . L'opérateur n'a aucune possibilité de modifier les conditions d'apparition du point critique, ce sont des constantes physiques du corps étudié.
Au point triple les trois phases sont en présence, soit :


Il n'y a aucun degré de liberté au point triple. Comme pour le point critique, l'opérateur ne peut modifier aucune des conditions d'apparition du point triple, ce sont également des constantes physiques du corps étudié.
Un corps pur ne peut être en équilibre sous plus de trois phases simultanément, la variance serait alors négative.
Équilibre liquide-vapeur binaire
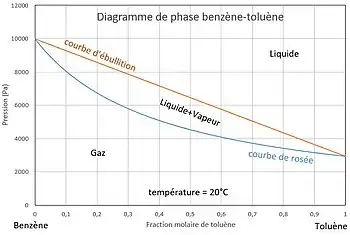
À l'équilibre liquide-vapeur, pour une température donnée, si l'on fixe la composition du liquide, la pression d'équilibre est subie (courbe d'ébullition) ainsi que la composition du gaz (courbe de rosée). Au-dessous de la courbe de rosée le gaz est seul, au-dessus de la courbe d'ébullition le liquide est seul : dans ces domaines on peut fixer, à température donnée, la pression indépendamment de la composition de la phase.
Pour un mélange binaire on a deux constituants, soit . On suppose qu'il n'est le siège d'aucune réaction chimique, soit . Si l'opérateur n'impose aucune condition opératoire, alors .

Pour le mélange sous forme de liquide seul ou de gaz seul, il n'y a qu'une seule phase, soit :

Il y a trois degrés de liberté. L'opérateur peut donc imposer la température, la pression et la concentration d'un composant indépendamment l'une de l'autre dans le domaine d'existence de la phase étudiée.
Lors des changements d'état on trouve deux phases en équilibre (liquide et gaz), soit :

Il y a deux degrés de liberté. Si l'opérateur fixe la température et la concentration de l'un des composants dans l'une des phases, la pression et la composition de l'autre phase sont subies. L'opérateur ne peut donc imposer à la fois la température, la pression et la composition de l'une des phases : avec on a .

Azéotrope
Un azéotrope présente une contrainte supplémentaire par rapport au cas précédent : les deux phases ont la même composition. La variance est donc diminuée d'un degré de liberté () ; la variance réduite vaut :

Si l'opérateur veut obtenir une composition particulière de l'azéotrope, afin de favoriser l'un des deux produits, il n'a pas le choix de la pression et de la température.
Allotropie et polymorphisme
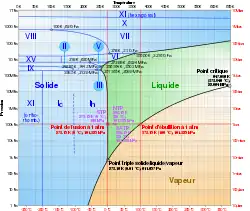
Un corps à l'état solide peut présenter diverses formes cristallines ou moléculaires : il s'agit de l'allotropie pour les corps simples et plus généralement du polymorphisme. Chacune des formes solides doit être comptée comme une phase à part entière dans la règle des phases.
Le diagramme ci-contre montre les domaines d'existence des différentes formes de la glace d'eau en fonction de la pression et de la température. Il existe un point triple à 100 K et 62 GPa. On compte :
- pour l'eau pure ;
- en l'absence de réaction ;
- en l'absence de contrainte ;
- pour la température et la pression ;
- (phases VII, VIII et X) ;
la variance vaut :
Il existe d'autres points triples impliquant trois solides. Le long de la courbe d'équilibre liquide-solide se trouvent plusieurs points triples impliquant deux phases solides et l'eau liquide. Comme pour le point triple solide-liquide-gaz, l'opérateur ne peut modifier les conditions de pression et température de tous ces points triples.
Combustion du soufre
La combustion du soufre solide S en présence d'oxygène O2 forme du dioxyde de soufre SO2 et du trioxyde de soufre SO3 en phase gaz selon deux réactions :


L'équilibre entre les deux oxydes n'est pas indépendant puisqu'il est une combinaison des deux réactions précédentes :



On a quatre constituants ( : S, SO2, SO3 et O2), deux réactions () et deux phases ( : solide et gaz). Si l'opérateur n'impose aucune condition opératoire, alors . La variance vaut :



L'opérateur peut imposer la pression d'oxygène et la température indépendamment l'une de l'autre selon le rendement cherché en l'un des deux produits SO2 et SO3.
Synthèse de l'ammoniac
Dans le procédé Haber-Bosch, l'ammoniac NH3 est synthétisé en phase gaz à partir d'azote N2 et d'hydrogène H2 selon la réaction :

On a (NH3, N2 et H2), , , et (gaz), d'où la variance :



L'opérateur peut fixer la pression, la température et la composition en azote ou en hydrogène selon le rendement en ammoniac cherché.
Décomposition de l'ammoniac
Par contre si l'on part de l'ammoniac pur, celui-ci se décompose selon la réaction inverse :

Il y a une contrainte supplémentaire par rapport au cas précédent : le mélange à l'équilibre contient trois fois plus d'hydrogène que d'azote. La variance est donc diminuée d'un degré de liberté ; la variance réduite vaut :

L'opérateur peut fixer la pression et la température indépendamment l'une de l'autre, la composition est alors subie. Il peut également se fixer la composition finale en ammoniac, de façon à éviter au maximum sa décomposition, et ajuster en conséquence la température (ou la pression) en subissant la pression (respectivement la température).
De façon générale, si les réactifs d'une réaction chimique sont introduits à la stœchiométrie, les produits seront également formés à la stœchiométrie. Ceci constitue donc une contrainte supplémentaire à inclure dans , puisque l'opérateur impose une condition opératoire.
Solubilité du sel de table
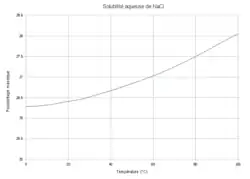
Sur la courbe, à saturation, si l'opérateur fixe une température la solubilité est subie, et réciproquement. Sous la courbe, en sous-saturation, l'opérateur peut imposer la température indépendamment de la concentration en sel dans la solution.
Le chlorure de sodium NaCl solide se dissout dans l'eau sous forme de deux ions : l'ion sodium Na+ et l'ion chlorure Cl−. La réaction s'écrit :

À l'équilibre réactionnel (état de saturation, la concentration en sel dissout est alors appelée solubilité du sel) on a :
- quatre constituants, soit (l'eau, NaCl, Na+ et Cl−) ;
- une réaction, soit ;
- une contrainte supplémentaire avec l'électroneutralité de la solution liquide qui impose que le nombre de charges positives (cations Na+) soit le même que celui de charges négatives (anions Cl−) ; on a donc, si l'opérateur ne fixe aucun paramètre, ;
- la présence de phases condensées uniquement (solide et liquide, pas de gaz), la pression n'a donc qu'une influence négligeable sur l'équilibre, on compte pour la température seule ;
- deux phases (un liquide et un solide, NaCl), soit .
La variance réduite vaut :

Il n'y a donc qu'un seul degré de liberté. Si l'opérateur fixe la température, la solubilité du sel est subie ; réciproquement, pour obtenir une certaine solubilité l'opérateur n'a pas le choix de la température.
En revanche, si la quantité de sel dissout est insuffisante pour atteindre la solubilité, la solution n'est pas saturée et le sel solide n'est pas présent : cet état est qualifié de sous-saturation. On a alors trois constituants ( : l'eau, Na+ et Cl−), pas de réaction (), l'électroneutralité () et une seule phase ( : le liquide) ; la variance réduite vaut :

En état de sous-saturation l'opérateur peut fixer indépendamment la température et la concentration en sel dissout, qui est alors inférieure à la solubilité à la même température. Cet état est un état d'équilibre, différent de l'équilibre à saturation car la phase solide n'existe pas, et la réaction n'a pas lieu.
Le passage de l'état de sous-saturation à celui de saturation, et inversement, est une rupture d'équilibre. Si l'on part de l'état de sous-saturation, donc en l'absence de NaCl solide, il y a déplacement d'équilibre lorsque l'on refroidit, ou lorsque l'on extrait de l'eau, tant que le sel reste entièrement dissout. Lorsque le sel précipité apparait la solution passe en état de saturation, il y a rupture d'équilibre : un nouvel état physico-chimique apparait en présence d'une nouvelle phase (solide), d'un nouveau constituant (NaCl) et d'une nouvelle réaction chimique (la précipitation). Inversement, si l'on part de l'état de saturation, donc en présence de NaCl solide, il y a déplacement d'équilibre lorsque l'on augmente la température, ou lorsque l'on ajoute de l'eau, tant que la phase solide subsiste : du sel se dissout progressivement. Lorsque le sel solide disparait la solution passe en état de sous-saturation, il y a rupture d'équilibre : un nouvel état physico-chimique apparait en l'absence de la phase solide, du constituant NaCl et de la réaction de précipitation.
Dissolution simultanée d'un autre sel avec un ion commun
On dissout simultanément au NaCl du sulfate de sodium Na2SO4, qui produit des ions sulfate SO2-
4 selon la réaction :

L'ion Na+ est commun aux deux sels NaCl et Na2SO4. Avec :
- (l'eau, NaCl, Na2SO4, Na+, Cl− et SO2-
4) ; - ;
- pour l'électroneutralité (le nombre de cations Na+ est égal à celui d'anions Cl− plus deux fois celui de SO2-
4) ; - ;
- (un liquide et deux solides, NaCl et Na2SO4) ;

la variance réduite vaut :

Si l'opérateur fixe la température les solubilités des deux sels sont subies. Si l'opérateur fixe la solubilité de l'un des sels, la solubilité de l'autre sel et la température sont subies.
Si le Na2SO4 est introduit en quantité insuffisante pour atteindre la saturation, il est entièrement dissout : il ne compte pas parmi les composants ( pour l'eau, NaCl, Na+, Cl− et SO2-
4), le seul équilibre est celui du NaCl (), l'électroneutralité est toujours vraie () ainsi que , et le seul solide est NaCl en présence du liquide (). La variance réduite vaut :


L'opérateur peut fixer indépendamment la température et la concentration en SO2-
4 ou Cl−. En effet, si l'opérateur ajoute du Na2SO4, l'ajout conséquent de Na+ fait précipiter le NaCl. Il peut ainsi contrôler la concentration de Cl− dans la solution.
On obtient si NaCl est également introduit en quantité insuffisante pour atteindre la saturation. L'opérateur peut fixer indépendamment la température et les concentrations en Cl− et SO2-
4.

Dissolution simultanée d'un autre sel sans ion commun
Au lieu de Na2SO4 on dissout en même temps que le NaCl du sulfate d'argent Ag2SO4, qui réagit selon :

Les deux sels NaCl et Ag2SO4 ne forment pas d'ion commun. Avec :
- (l'eau, NaCl, Ag2SO4, Na+, Ag+, Cl− et SO2-
4) ; - ;
- ; on a trois contraintes supplémentaires dont deux seulement sont indépendantes :
- le nombre d'ions Na+ égale celui de Cl− ;
- le nombre d'ions Ag+ est le double de celui de SO2-
4 ; - l'électroneutralité est composée des deux contraintes précédentes (le nombre de cations Na+ plus celui de Ag+ est égal au nombre d'anions Cl− plus deux fois celui de SO2-
4) ;
- ;
- (un liquide et deux solides, NaCl et Ag2SO4) ;

la variance réduite vaut :

Comme pour le cas avec ion commun, si l'opérateur fixe la température les solubilités des deux sels sont subies. Si l'opérateur fixe la solubilité de l'un des sels, la solubilité de l'autre sel et la température sont subies.
On obtient si Ag2SO4 est introduit en quantité insuffisante pour atteindre la saturation. La variance est identique au cas avec ion commun, cependant ici l'ajout de Ag2SO4 n'a aucune influence sur la quantité de NaCl dissout car les deux réactions sont indépendantes. L'opérateur ne peut modifier indépendamment que la température et la teneur en Ag2SO4 dissout, la quantité de NaCl dissout est subie.
Si le NaCl est également en sous-saturation, on a . Les conclusions sont identiques à celles du cas avec ion commun : on peut faire varier indépendamment la température et les deux concentrations des sels dissouts.
Notes et références
Notes
- Jean Hertz, « Historique en grandes enjambées de la thermodynamique de l’équilibre », J. Phys. IV France, vol. 122, , p. 3-20 (lire en ligne [PDF], consulté le ).
- Exemple de sept phases liquides (heptane, aniline, eau, perfluorokérosène, phosphore, gallium, mercure) en équilibre à pression et température ambiantes : (en) Joel H. Hildebrand, « Seven Liquid Phases in Equilibrium. », Journal of Physical Chemistry, vol. 53, no 6, , p. 944-947 (lire en ligne).
- Odile Durupthy, Alain Jaubert, André Durupthy et Jacques Estienne, Chimie 2e année MP-MP*/PT-PT* : Cours avec exercices corrigés, Hachette Éducation, coll. « H Prépa », , 256 p. (ISBN 978-2-01-181760-0, lire en ligne), p. 80.
Bibliographie
- Livres
- Jean-Claude Legrand, L'indispensable en Thermodynamique chimique : les applications, Bréal, (ISBN 978-2-7495-2241-8, lire en ligne), p. 15-17.
- J. Mesplède, Chimie : Thermodynamique Matériaux PC, Bréal (ISBN 978-2-7495-2064-3, lire en ligne), p. 86-90.
- Richard Taillet, Loïc Villain et Pascal Febvre, Dictionnaire de physique : + de 6500 termes, nombreuses références historiques, des milliers de références bibliographiques, Louvain-la-Neuve, De Boeck supérieur, , 976 p. (ISBN 978-2-8073-0744-5, lire en ligne), p. 559.
- Michel Soustelle, Équilibres chimiques, vol. 4, Londres, ISTE Group, coll. « Génie des procédés / Thermodynamique chimique approfondie », , 196 p. (ISBN 978-1-78405-103-7, lire en ligne), p. 82-85 ; p. 93.
- Article
- Mohamed Jemal, « Sur le calcul de la variance », L'actualité de la chimie, no 333, , p. 38-41 (lire en ligne [PDF], consulté le ).
Liens externes
- « Variance d'un système en équilibre », sur sciences-en-ligne.com (consulté le ).