Loi de Raoult
En physique, et plus particulièrement en thermodynamique, la loi de Raoult énonce que :
« Dans une solution idéale, à température constante, la pression partielle en phase vapeur d'un constituant est proportionnelle à sa fraction molaire en phase liquide. »
Cette loi a été établie empiriquement par le physicien français François-Marie Raoult en 1882, elle est dérivée de sa loi de la tonométrie. Elle est utilisée dans de nombreux domaines de la chimie, de la physique et de la météorologie. Elle permet notamment de calculer les équilibres liquide-vapeur des solutions idéales liquides dont la phase vapeur est un mélange de gaz parfaits.
Énoncé et démonstration
Énoncé mathématique
Étant donné que pour le corps pur la pression est égale à sa pression de vapeur saturante, le coefficient de proportionnalité est égal à cette pression à la température du mélange. La forme mathématique de la loi de Raoult s'écrit, pour une espèce chimique :

| Loi de Raoult pression partielle de : |

avec les notations :
- la pression totale du mélange ;
- la pression partielle du composant , par définition ;
- la pression de vapeur saturante du composant pur à la température du mélange ;
- la fraction molaire du constituant dans la phase vapeur ;
- la fraction molaire du constituant dans la phase liquide.







Pour un corps pur, et par conséquent .


Dans un mélange, la relation de Duhem-Margules impose que si l'un des composants suit la loi de Raoult alors les autres composants suivent également la loi de Raoult, ou, pour les gaz dissouts, la loi de Henry.
Démonstration
La loi de Raoult a été établie empiriquement par son auteur, sur base de sa loi de la tonométrie pour les liquides et de la loi de Dalton pour les gaz. La démonstration rigoureuse qui suit est postérieure au développement du formalisme mathématique de la thermodynamique par Willard Gibbs, qui introduisit notamment la notion de potentiel chimique, et par Gilbert Lewis, qui introduisit les notions de fugacité et d'activité chimique.
Dans une solution à l'équilibre, pour chaque constituant les potentiels chimiques dans la phase liquide et dans la phase vapeur sont égaux :


- (1)

Pour la phase liquide et la phase vapeur, considérées comme des solutions idéales, les potentiels chimiques sont développés selon :
- (2)
- (3)


avec :
- la pression totale du mélange ;
- la constante universelle des gaz parfaits ;
- la température.

La variation isotherme du potentiel chimique en fonction de la pression permet d'écrire pour le composant pur en phase liquide :

avec volume molaire du composant à l'état de liquide pur. Le dernier terme est généralement négligé, soit :


Ceci traduit le fait que les liquides sont généralement peu sensibles à la pression et sont considérés comme incompressibles : leurs propriétés sont considérées, à température donnée, comme étant celles du liquide à saturation à cette même température, quelle que soit la pression. En introduisant cette relation dans (2), il vient :
- (4)

La variation isotherme du potentiel chimique en fonction de la pression permet d'écrire pour le composant pur en phase vapeur :

avec le volume molaire du composant à l'état de gaz pur. Si ce gaz est considéré comme parfait alors :



En introduisant cette relation dans (3), il vient :
- (5)

Les relations (4) et (5) permettent donc de réécrire la relation (1) selon :
- (6)

La relation (1) étant aussi valable pour un corps pur à l'équilibre liquide-vapeur (saturation), celui-ci à la température étant à sa pression de vapeur saturante , il vient :

La relation (6) produit donc :

Correction de Poynting
Si l'on ne néglige pas le terme dans l'expression du potentiel chimique en phase liquide, celui-ci dans la relation (4) s'exprime exactement selon :



avec volume molaire du composant liquide pur à et . Le facteur exponentiel est appelé correction de Poynting ou facteur de Poynting :


La loi de Raoult avec correction de Poynting devient :
| Loi de Raoult et correction de Poynting pression partielle de : |

Le liquide pur n'existe de façon stable à la température que pour des pressions . Pour des pressions le liquide pur soit n'existe pas, soit existe sous une forme thermodynamiquement instable : on peut alors ne pas trouver de valeur pour . Toutefois, les liquides étant peu compressibles, le volume molaire liquide peut être considéré comme indépendant de la pression ; il est souvent donné à saturation du liquide : , auquel cas :




Ceci permet de calculer la correction de Poynting à toute pression, y compris celles pour lesquelles, à température donnée, le corps pur n'existe pas sous forme de liquide stable.
Aux basses pressions (moins de 10 atm, domaine d'application de la loi des gaz parfaits), la correction de Poynting est négligeable :

Pour un mélange, la relation de Duhem-Margules induit que si l'un des composants suit la loi de Raoult avec correction de Poynting alors les autres composants suivent également la loi de Raoult avec correction de Poynting ou la loi de Henry avec correction de Poynting.
Avec la loi de Raoult sans correction de Poynting, connaissant pour un mélange liquide sa composition (fractions molaires ) et la pression de vapeur saturante de chacun des composants à la température , la pression se calcule explicitement selon :

La loi de Raoult avec correction de Poynting nécessite un calcul itératif, puisque l'expression est implicite en :

Limites et extension de la loi de Raoult
Écarts des équilibres liquide-vapeur réels
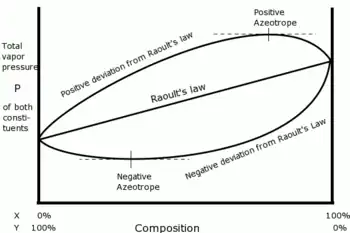
La loi de Raoult s’applique à des systèmes idéaux dans lesquels les interactions entre molécules de même espèce ou d'espèces différentes sont toutes identiques. Les écarts à la loi de Raoult observés dans les équilibres liquide-vapeur réels peuvent être positifs ou négatifs selon l'attraction électronique des composants du mélange.
- Écart positif
- Lorsque dans un mélange composé de deux constituants A et B les molécules du même constituant (interactions A-A et B-B) s'attirent plus fortement que les molécules d'espèces différentes entre elles (interaction A-B), l’ajout par exemple de molécules B dans le liquide A pur a pour effet de diminuer les forces d’attraction entre les molécules A et la volatilité de A est augmentée. Il y a alors une déviation positive par rapport à la loi de Raoult : la pression totale de la vapeur en équilibre au-dessus de la solution liquide est supérieure à celle calculée.
- Écart négatif
- Si, par contre, les molécules de A et de B sont plus attirées entre elles (interaction A-B) que par celles du même constituant (interactions A-A et B-B), l'ajout de molécules B au liquide A pur augmente les forces d’attraction auxquelles sont soumises les molécules A et celles-ci deviennent moins volatiles. Il y a alors une déviation négative par rapport à la loi de Raoult : la pression totale de la vapeur en équilibre au-dessus de la solution liquide est inférieure à celle calculée.
Le plus souvent les pressions d'équilibre d'un mélange binaire réel restent comprises, pour une température donnée, entre les pressions de vapeur saturante des deux composants à cette même température.
Les azéotropes
Lorsque le comportement non idéal d'un mélange binaire devient suffisamment important, les pressions d'équilibre du mélange peuvent être soit plus grandes que la plus grande des pressions de vapeur saturante des composants, soit plus petites que la plus petite des pressions de vapeur saturante des composants, ce qui est représenté sur la figure ci-dessus. Dans les deux cas les courbes d'équilibre liquide-vapeur présentent nécessairement un extremum (respectivement un maximum et un minimum) qui correspond, en vertu du théorème de Gibbs-Konovalov, à un azéotrope. L'azéotrope est dit positif si sa pression est supérieure à la pression prévue par la loi de Raoult et négatif dans le cas contraire.
La loi de Raoult est fondamentalement incapable de représenter des azéotropes, c'est-à-dire des mélanges pour lesquels, dans certaines conditions de pression, température et composition, le liquide et la vapeur en équilibre ont la même composition. Dans ces conditions la pression d'ébullition et la pression de rosée (voir plus bas) sont égales : un mélange azéotropique se comporte donc comme un corps pur, néanmoins il y a bien plusieurs espèces chimiques en présence. Par exemple, le mélange eau - éthanol contenant 96 % d'éthanol présente un azéotrope à 78,1 °C sous une pression de 1 atm : la loi de Raoult ne peut pas représenter ce mélange.
Un azéotrope est une preuve de non-idéalité du mélange. Dans le cas du mélange eau - éthanol, cette non-idéalité se traduit également par le fait qu'un mélange d'un litre d'eau et d'un litre d'éthanol a un volume de 1,92 litre : les forces d'attraction entre molécules des deux espèces produisent une contraction du volume. La loi de Raoult étant basée sur l'hypothèse de l'idéalité des phases liquide et vapeur, ce type de comportement est donc en dehors de son champ d'application.
Soit un mélange binaire, contenant les espèces 1 et 2, présentant un azéotrope. On note :
- et les fractions molaires en phase vapeur ;
- et les fractions molaires en phase liquide ;
- et les pressions de vapeur saturante à la température ;
- la pression d'ébullition (voir définition plus bas) à la température ;
- la pression de rosée (voir définition plus bas) à la température .








On cherche la composition de l'azéotrope, point auquel , et .



La pression d'ébullition est calculée par (voir le paragraphe Diagrammes de phase binaires idéaux) :

on a :

La pression de rosée est calculée par (voir le paragraphe Diagrammes de phase binaires idéaux) :

on a :

Au point d'azéotrope, on a :



soit :

Cette équation n'admet que deux solutions :
- , auquel cas et , le corps 1 est pur ;
- , auquel cas et , le corps 2 est pur.






La loi de Raoult est donc incapable de représenter un azéotrope.
Cas des gaz dissouts, loi de Henry
La loi de Raoult s'applique à des composants qui présentent une pression de vapeur saturante à la température du mélange. Pour un gaz dissout, c'est-à-dire un fluide supercritique dont la température critique est inférieure à la température du mélange, soit , la loi de Raoult n'est pas applicable, puisque le gaz ne possède pas de pression de vapeur saturante à cette température. Pour les gaz dissouts, le pendant de la loi de Raoult est la loi de Henry.

La loi de Henry établit la relation suivante entre la pression partielle d'un constituant gazeux et sa fraction molaire dans un solvant liquide :


où est appelé constante de Henry, bien qu'elle dépende de la température et de la pression. La constante de Henry est spécifique du gaz dans le solvant , c'est-à-dire qu'elle doit être déterminée pour chaque couple « gaz - solvant » et n'est pas valable si le gaz est considéré avec un solvant autre que le solvant pour lequel elle a été déterminée.

Extension aux équilibres liquide-vapeur réels
La loi de Raoult n'est valable que si le liquide et sa vapeur sont tous deux des solutions idéales, la phase gaz étant de surcroît un mélange de gaz parfaits. L'application de la loi de Raoult est donc restreinte à des cas particuliers, pour certains mélanges de produits et certaines plages de température et de pression. Elle constitue néanmoins une base pour calculer les équilibres liquide-vapeur des mélanges réels (y compris les azéotropes) à l'aide de facteurs correctifs tels que les coefficients de fugacité pour la phase gaz et les coefficients d'activité pour la phase liquide :

avec :
- le coefficient de fugacité du corps en phase gaz, à , et composition du mélange gazeux ;
- le coefficient de fugacité du corps pur à saturation en phase gaz, à et ;
- le coefficient d'activité du corps en phase liquide, à , et composition du mélange liquide ;
- la correction de Poynting.




Applications
Pression d'ébullition
La pression d'ébullition d'un mélange, à température donnée, est la pression au-delà de laquelle (pour des pressions plus fortes) le mélange est entièrement liquide : à la pression d'ébullition apparaît la première bulle de vapeur. En deçà de la pression d'ébullition (pour des pressions plus faibles), le mélange présente un équilibre liquide-vapeur, ou est entièrement gazeux (en deçà de la pression de rosée). Soit un mélange liquide de composants, chaque composant étant représenté par sa fraction molaire , à la température .

Nous connaissons la température du mélange, nous pouvons donc calculer la pression de vapeur saturante de chacun des composants. La composition de la phase liquide est connue. On suppose que tous les composants du mélange suivent la loi de Raoult. La loi de Raoult sans correction de Poynting donne, en sommant sur l'ensemble des constituants :

La pression d'ébullition se calcule donc selon[1] - [2] :
| Pression d'ébullition : |

Toutefois avec la correction de Poynting le calcul n'est pas explicite. En effet, l'équation à résoudre en devient :


On pourra employer la méthode du point fixe[1]. Soit la fonction de :


On initialise le calcul par la solution obtenue par la loi de Raoult sans correction de Poynting :

puis on calcule la pression à partir de la pression obtenue à l'itération précédente par :



jusqu'à une erreur acceptable :


Exemple[3]
- On considère un mélange d'hydrocarbures aromatiques dans les conditions données par le tableau suivant.
| Constituant | Fraction molaire liquide | Pression de vapeur saturante en mmHg |
|---|---|---|
| Benzène | 0,4 | 271,29 |
| Toluène | 0,4 | 92,11 |
| Éthylbenzène | 0,2 | 35,16 |
- La pression de bulle est de .

Température d'ébullition
La température d'ébullition d'un mélange, à pression donnée, est la température en deçà de laquelle (pour des températures plus basses) le mélange est entièrement liquide : à la température d'ébullition apparaît la première bulle de vapeur. Au-delà de la température d'ébullition (pour des températures plus élevées), le mélange présente un équilibre liquide-vapeur, ou est entièrement gazeux (au-delà de la température de rosée). Soit un mélange liquide de composants, chaque composant étant représenté par sa fraction molaire , à la pression .
La solution n'est pas explicite, car l'expression de la loi de Raoult (avec ou sans correction de Poynting) n'est pas explicite en température. La pression étant donnée et la température étant l'inconnue, les pressions de vapeur saturante et les corrections de Poynting sont des fonctions de . Si l'on pose la fonction de la température :



la température d'ébullition est la solution de l'équation : , la pression étant une donnée du problème. On pourra travailler par itérations imbriquées, en faisant varier la température et en calculant la pression d'ébullition correspondante jusqu'à ce que cette pression soit la pression de l'énoncé[1] - [2].


Pression de rosée
La pression de rosée d'un mélange, à température donnée, est la pression en deçà de laquelle (pour des pressions plus faibles) le mélange est entièrement gazeux : à la pression de rosée apparaît la première goutte de liquide. Au-delà de la pression de rosée (pour des pressions plus fortes), le mélange présente un équilibre liquide-vapeur, ou est entièrement liquide (au-delà de la pression d'ébullition). Soit un mélange gazeux de composants, chaque composant étant représenté par sa fraction molaire , à la température .
Nous connaissons la température du mélange, nous pouvons donc calculer la pression de vapeur saturante de chacun des composants. La composition de la phase vapeur est connue. On suppose que tous les composants du mélange suivent la loi de Raoult. La loi de Raoult sans correction de Poynting donne pour tout constituant :

et, en sommant sur l'ensemble des constituants :

La pression de rosée se calcule donc selon[4] - [5] :
| Pression de rosée : |

Toutefois avec la correction de Poynting le calcul n'est pas explicite. En effet, l'équation à résoudre en devient :

On pourra employer la méthode du point fixe[4]. Soit la fonction de :

On initialise le calcul par la solution obtenue par la loi de Raoult sans correction de Poynting :

puis on calcule la pression à partir de la pression obtenue à l'itération précédente par :

jusqu'à une erreur acceptable :
Exemple[3]
- On considère un mélange d'hydrocarbures aromatiques dans les conditions données par le tableau suivant.
| Constituant | Fraction molaire vapeur | Pression de vapeur saturante en mmHg |
|---|---|---|
| Benzène | 0,4 | 271,29 |
| Toluène | 0,4 | 92,11 |
| Éthylbenzène | 0,2 | 35,16 |
- La pression de rosée est de .

Température de rosée
La température de rosée d'un mélange, à pression donnée, est la température au-delà de laquelle (pour des températures plus élevées) le mélange est entièrement gazeux : à la température de rosée apparaît la première goutte de liquide. En deçà de la température de rosée (pour des températures plus basses), le mélange présente un équilibre liquide-vapeur, ou est entièrement liquide (en deçà de la température d'ébullition). Soit un mélange gazeux de composants, chaque composant étant représenté par sa fraction molaire , à la pression .
La solution n'est pas explicite, car l'expression de la loi de Raoult (avec ou sans correction de Poynting) n'est pas explicite en température. La pression étant donnée et la température étant l'inconnue, les pressions de vapeur saturante et les corrections de Poynting sont des fonctions de . Si l'on pose la fonction de la température :

la température de rosée est la solution de l'équation : , la pression étant une donnée du problème. On pourra travailler par itérations imbriquées, en faisant varier la température et en calculant la pression de rosée correspondante jusqu'à ce que cette pression soit la pression de l'énoncé[6] - [2].


Énoncé du problème
Soit, à pression et température données, un mélange de constituants, chaque constituant étant représenté par la quantité . On cherche à déterminer la composition des phases vapeur et liquide en présence.

Nous avons inconnues, il s'agit en effet de calculer pour chacun des constituants :

- la quantité du constituant en phase vapeur ;
- la quantité du constituant en phase liquide.


Soient :
- la quantité de matière totale en phase vapeur (inconnue) ;
- la quantité de matière totale en phase liquide (inconnue) ;
- la fraction molaire du constituant en phase vapeur (inconnue) ;
- la fraction molaire du constituant en phase liquide (inconnue).




On suppose que tous les composants du mélange suivent la loi de Raoult. La pression et la température étant données, les pressions de vapeur saturante et les corrections de Poynting sont des constantes. On définit pour chacun des constituants le coefficient de partage , qui est une constante en appliquant la loi de Raoult à pression et température données :


Nous avons les équations :
- équilibres chimiques selon la loi de Raoult : ;
- bilans de matière : .


Réduction du problème
Soit la quantité de matière totale considérée, il s'agit d'une valeur connue du problème. On a, par bilan de matière global :


Soit pour chacun des constituants la fraction molaire globale, notée :


Puisque l'on connaît la quantité de matière de chacun des constituants, les fractions molaires sont connues également.
On définit d'autre part la fraction molaire globale de la phase vapeur, ou taux de vaporisation :


Cette grandeur est une inconnue du problème. Le système de équations à inconnues est réduit à une équation à une inconnue, , due à Rachford et Rice (1952)[7] - [6] - [8] - [9] :
| Équation de Rachford-Rice : |

On a par définition des fractions molaires dans les deux phases :


En introduisant ces relations dans le bilan de matière de chacun des constituants :

et en divisant par on obtient pour chacun des constituants :


En substituant dans cette dernière relation l'équation d'équilibre chimique du constituant :

On remarquera que si , alors : le constituant n'est pas présent dans le mélange ; ce cas est donc exclu. On peut donc écrire[9] :



d'où également, toujours d'après la relation de l'équilibre chimique :

Dans les expressions des fractions et ainsi établies, seul le taux de vaporisation est inconnu. Nous pouvons donc réduire le problème à la recherche de cette seule inconnue. Soit la fonction de définie par[9] :

Cette fonction est monotone strictement décroissante en pour . Si l'on trouve sur cette plage une solution à l'équation , elle est unique et .



Il reste à prouver que cette solution respecte les contraintes sur les fractions molaires, soit et . Revenons à la relation pour chacun des constituants :


En sommant sur l'ensemble des constituants :

or puisque , alors :

Par construction , d'où et .

On pourra résoudre cette équation sur l'intervalle par la méthode de Newton, en considérant la dérivée de la fonction :



On remarquera que , ce qui indique que est décroissante.

On initialisera le calcul avec , puis on procèdera par itérations successives selon[9] :


jusqu'à une erreur acceptable :

Les fractions molaires sont ensuite calculées par[9] :
et les quantités globales respectives des deux phases par :


d'où l'on obtient finalement les quantités dans chaque phase pour chacun des constituants :
- Note - autre forme de l'équation de Rachford-Rice
- Plutôt que de rechercher le taux de vaporisation , on peut rechercher le taux de liquéfaction défini par :


- L'équation de Rachford-Rice se présente alors sous la forme :

- Cette fonction est monotone strictement croissante en pour .

Résolution du problème
Avant de calculer un équilibre liquide-vapeur, il est nécessaire de vérifier que le mélange n'est pas monophasique, soit uniquement liquide ou gazeux.
À température donnée, le coefficient de partage de chacun des constituants est une fonction de la pression :

Lorsque la pression est supérieure à la pression d'ébullition, soit , le mélange est entièrement liquide. Pour une composition donnée, la fonction est une fonction décroissante de la pression. Lorsque la pression est la pression d'ébullition aux température et composition données. Lorsque on a [10].




Lorsque la pression est inférieure à la pression de rosée, soit , le mélange est entièrement gazeux. Pour une composition donnée, la fonction est une fonction croissante de la pression. Lorsque la pression est la pression de rosée aux température et composition données. Lorsque on a [10].




- Algorithme de calcul
On effectuera par conséquent le calcul selon l'algorithme suivant :
- à pression, température et composition de l'énoncé calculer la fraction molaire globale et le coefficient de partage pour chacun des constituants ;
- si le mélange est entièrement liquide, et pour tout constituant , fin du calcul ;
- si le mélange est entièrement gazeux, et pour tout constituant , fin du calcul ;
- sinon, le mélange est biphasique et présente un équilibre liquide-vapeur, résoudre l'équation de Rachford-Rice : .







Dans le cas d'un mélange binaire, impliquant les constituants et , après avoir vérifié la présence de deux phases comme précédemment, on calcule plus rapidement les compositions des phases selon[11] :


- ;
- ;
- ;
- ;
- .





Exemple[12]
- On considère un mélange d'hydrocarbures dans les conditions données par le tableau suivant.
| Constituant | Fraction molaire globale | Coefficient de partage |
|---|---|---|
| Propane | 0,2 | 2,525 |
| n-Butane | 0,3 | 0,7708 |
| Isobutane | 0,4 | 1,066 |
| n-Pentane | 0,05 | 0,2401 |
| Isopentane | 0,05 | 0,3140 |
- On a et . Le mélange est donc biphasique. Les résultats sont reportés dans le tableau suivant.


| Taux de vaporisation | 0,524 | |
|---|---|---|
| Constituant | Fraction molaire liquide | Fraction molaire vapeur |
| Propane | 0,1112 | 0,2807 |
| n-Butane | 0,3409 | 0,2628 |
| Isobutane | 0,3867 | 0,4121 |
| n-Pentane | 0,0831 | 0,0199 |
| Isopentane | 0,0781 | 0,0245 |
Principe
La relation de Duhem-Margules induit que dans un mélange binaire si l'un des corps suit la loi de Raoult alors le deuxième suit également la loi de Raoult (ou la loi de Henry). On considère par conséquent deux corps et suivant la loi de Raoult à l'équilibre liquide-vapeur. On a six inconnues :
- la température ;
- la pression ;
- la fraction molaire du corps en phase vapeur ;
- la fraction molaire du corps en phase liquide ;
- la fraction molaire du corps en phase vapeur ;
- la fraction molaire du corps en phase liquide ;




et quatre équations :
- la loi de Raoult pour le corps : ;
- la loi de Raoult pour le corps : ;
- la contrainte sur les fractions molaires de la phase vapeur : ;
- la contrainte sur les fractions molaires de la phase liquide : .




Les pressions de vapeur saturante et sont des fonctions de la température.


On a donc deux degrés de liberté, conformément à la règle des phases : on peut étudier l'équilibre liquide-vapeur de ce système en fixant deux paramètres parmi les six inconnues définies plus haut. Par exemple, en fixant la température (diagrammes isothermes), on peut :
- fixer la composition du liquide et calculer la pression : on obtient le point d'ébullition ;
- fixer la composition du gaz et calculer la pression : on obtient le point de rosée ;
- fixer la composition du liquide et calculer la composition du gaz (et inversement).
On peut de façon similaire fixer la pression (diagrammes isobares) et la composition de l'une des phases pour calculer la température d'équilibre ou la composition de l'autre phase.
Diagrammes isothermes
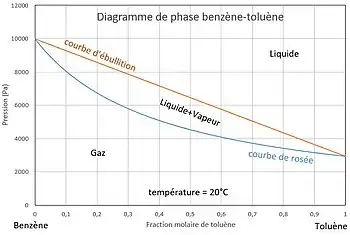
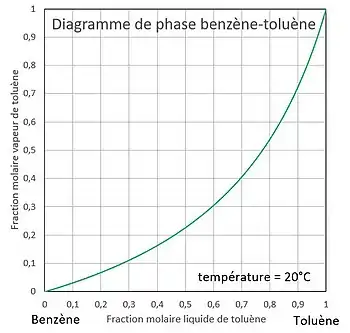
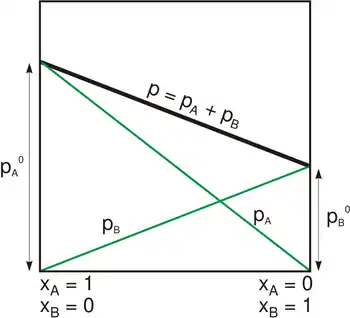
La température est fixée. On veut tracer l'évolution de la pression en fonction de la composition.
Puisque la température est fixée, les pressions de vapeur saturante et sont constantes.
En considérant la contrainte sur la composition de la vapeur et la loi de Raoult :


Si l'on choisit, arbitrairement, le corps comme référence, avec la contrainte sur la composition liquide on a :

On obtient la formule de la courbe d'ébullition :

En considérant la contrainte sur la composition du liquide et la loi de Raoult :


Si l'on choisit, arbitrairement, le corps comme référence, avec la contrainte sur la composition vapeur on a :

On obtient la formule de la courbe de rosée :

La courbe d'ébullition est un segment de droite. La courbe de rosée est un arc d'hyperbole. Les deux courbes ne peuvent donc s'intercepter qu'en deux points, ces points correspondent aux points d'ébullition des deux corps purs, soit et . La loi de Raoult est ainsi incapable de représenter un azéotrope, point auquel le liquide et la vapeur d'un mélange binaire ont la même composition.


Il est également possible de tracer la courbe de composition d'une phase en fonction de la composition de l'autre phase, par exemple de la vapeur en fonction du liquide : . En effet, à la température fixée la pression est donnée par les deux relations :


On obtient par conséquent :

Diagrammes isobares
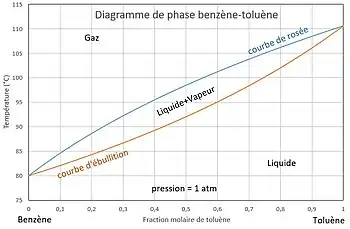
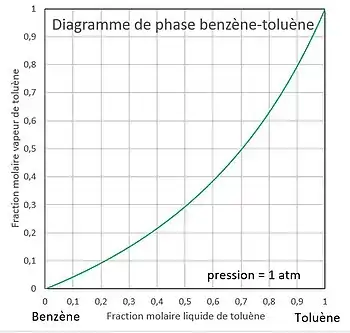
La pression est fixée. On veut tracer l'évolution de la température en fonction de la composition.
Les pressions de vapeur saturante et sont des fonctions de la température. Il est donc nécessaire de disposer de corrélations permettant cette évaluation. En intégrant la formule de Clausius-Clapeyron, la pression de vapeur saturante d'un corps pur peut être écrite, par exemple, selon la formule de Rankine :
![{\displaystyle \ln \!\left({P_{i}^{\text{sat}} \over P}\right)=-{\Delta _{\text{vap}}H_{i} \over R}\left[{1 \over T}-{1 \over T_{{\text{éb}},i}}\right]}](https://img.franco.wiki/i/4286e48ec8eea7b8e37706532e6c56045991aeef.svg)
avec :
- la température d'ébullition du corps pur sous la pression , il s'agit d'une constante ;
- la pression de vapeur saturante du corps pur à la température ; il s'agit donc d'une fonction telle que , et en particulier ;
- l'enthalpie de vaporisation du corps pur à la température (donc sous la pression ), supposée constante dans l'établissement de la formule de Rankine (l'enthalpie de vaporisation dépend normalement de la température).



On pose les coefficients de partage :


En considérant la contrainte sur la composition de la vapeur et la loi de Raoult :

Avec la contrainte sur la composition du liquide on a :

d'où :

Avec la loi de Raoult on obtient :


Ainsi, en imposant la température , la pression étant donnée, on peut calculer et , puis , , et . Si l'on choisit, arbitrairement, le corps comme référence, en faisant varier la température entre et , la courbe est la courbe d'ébullition, la courbe est la courbe de rosée.






Il est également possible de tracer la courbe de composition d'une phase en fonction de la composition de l'autre phase, par exemple de la vapeur en fonction du liquide : . Il suffit, à la pression fixée :
- de poser ,
- de trouver entre et telle que ,
- de calculer .

Notes et références
Notes
- Vidal 1997, p. 198.
- Shell 2015, p. 250.
- (en) Kevin D. Dahm et Donald P. Visco, Fundamentals of Chemical Engineering Thermodynamics, Cengage Learning, , 800 p. (ISBN 9781111580704, lire en ligne), p. 458-459.
- Vidal 1997, p. 199.
- Shell 2015, p. 251.
- Vidal 1997, p. 200.
- (en) H.H. Rachford et J.D. Rice, « Procedure for Use of Electronic Digital Computers in Calculating Flash Vaporization Hydrocarbon Equilibrium », Journal of Petroleum Technology, vol. 4, no 10, , p. 19-20 (DOI https://doi.org/10.2118/952327-G, lire en ligne [PDF], consulté le ).
- Shell 2015, p. 252.
- Christophe Coquelet et Dominique Richon, Propriétés thermodynamiques : Détermination pour les mélanges, vol. BE 8031, Techniques de l'ingénieur, coll. « base documentaire : Thermodynamique et énergétique, pack : Physique énergétique, univers : Énergies », , 12 p., p. 8-9.
- Shell 2015, p. 253.
- Vidal 1997, p. 204.
- Vidal 1997, p. 203.
Bibliographie
- Articles
- E. Darmois, « La thermodynamique des solutions », J. Phys. Radium, vol. 4, no 7, , p. 129-142 (lire en ligne, consulté le ).
- Publications
- J. Mesplède, Chimie : Thermodynamique Matériaux PC : Cours, méthode, exercices résolus, Éditions Bréal, coll. « Les nouveaux précis Bréal », (ISBN 978-2-7495-2064-3, lire en ligne), p. 124-128.
- Jean Vidal, Thermodynamique : application au génie chimique et à l'industrie pétrolière, Paris, Éditions Technip, coll. « Publications de l'Institut français du pétrole. », , 500 p. (ISBN 978-2-7108-0715-5, OCLC 300489419, lire en ligne), p. 184, 197-205.
- (en) M. Scott Shell, Thermodynamics and Statistical Mechanics : An Integrated Approach, Cambridge University Press, , 476 p. (ISBN 9781107014534, lire en ligne), p. 220-221, 250-253.
- (en) Robert C. Reid, John M. Prausnitz et Bruce E. Poling, The properties of gases and liquids, New York, McGraw-Hill, , 4e éd., 741 p. (ISBN 978-0-07-051799-8).
Liens externes
- Claude Anies, Institut privé de préparation aux études supérieures, « Chapitre V : Diagramme binaire liquide-vapeur », sur KlubPrépa (consulté le ).