En chimie physique, et plus particulièrement en thermodynamique, la relation de Duhem-Margules, appelée également relation de Gibbs-Duhem-Margules ou équation de Duhem-Margules, lie les variations des pressions partielles d'une phase vapeur aux variations de la composition d'une phase liquide en équilibre à température constante.
Cette relation a d'abord été établie par Willard Gibbs en 1876, puis indépendamment par Pierre Duhem en 1886, R. A. Lehfeldt[1] en 1895 et Max Margules la même année[2] - [3].
La relation de Duhem-Margules est démontrée en supposant une phase vapeur idéale, c'est-à-dire se comportant comme un mélange de gaz parfaits. Il n'est par contre fait aucune hypothèse sur la nature de la phase liquide, qui peut en conséquence ne pas être idéale. Ainsi la relation de Duhem-Margules représente-t-elle correctement les équilibres liquide-vapeur réels aux pressions proches de la pression atmosphérique, y compris donc ceux qui ne suivent pas la loi de Raoult, voire ceux qui présentent un azéotrope.
Elle n'est cependant pas rigoureuse, car elle est établie en supposant que, à température constante, la pression totale de la vapeur ne varie pas en fonction de la composition du liquide, ce qui est en contradiction avec la règle des phases. Aux basses pressions il est toutefois possible de justifier cette approximation[4].
La relation de Duhem-Margules permet de démontrer les règles de Konovalov. Elle a des implications sur la loi de Raoult et la loi de Henry. Margules en a également tiré son modèle de coefficients d'activité.
Énoncé et démonstration
Énoncé de la relation
Soit un mélange liquide binaire, constitué des corps  et
et  , en équilibre avec une phase vapeur se comportant comme un mélange de gaz parfaits. À température constante, les pressions partielles des deux corps en phase vapeur varient en fonction de la composition du liquide et sont liées par la relation de Duhem-Margules :
, en équilibre avec une phase vapeur se comportant comme un mélange de gaz parfaits. À température constante, les pressions partielles des deux corps en phase vapeur varient en fonction de la composition du liquide et sont liées par la relation de Duhem-Margules :
Relation de Duhem-Margules :  |
avec :
 la température ;
la température ; et
et  les pressions partielles respectives des corps et en phase vapeur ;
les pressions partielles respectives des corps et en phase vapeur ; et
et  les fractions molaires respectives des corps et en phase liquide.
les fractions molaires respectives des corps et en phase liquide.
Cette relation se trouve également sous les formes :
Relation de Duhem-Margules


Hypothèse de la pression constante
On trouve dans la littérature la relation de Duhem-Margules énoncée à pression  et température constantes[5] - [4] :
et température constantes[5] - [4] :

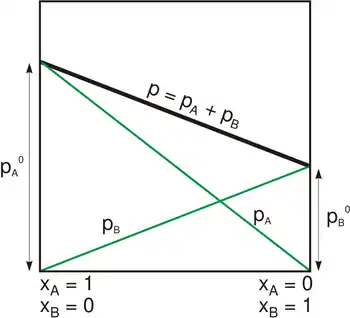
Exemple d'évolution à température constante des pressions totale et partielles selon la
loi de Raoult. La pression de vapeur totale (en noir) d'un mélange binaire est la somme des pressions partielles des deux corps (en vert) calculées en fonction des concentrations molaires de ceux-ci dans le mélange liquide. À température constante, la pression totale d'équilibre varie en fonction de la composition du liquide.
Or il est impossible de faire varier la composition d'un liquide binaire tout en maintenant un équilibre liquide-vapeur à pression et température constantes. En effet, selon la règle des phases énoncée par Gibbs, un mélange de deux constituants possède  degrés de liberté ; or, avec :
degrés de liberté ; or, avec :
 le nombre de constituants ;
le nombre de constituants ; le nombre de contraintes expérimentales, puisque et sont imposées ;
le nombre de contraintes expérimentales, puisque et sont imposées ; le nombre de phases en équilibre ;
le nombre de phases en équilibre ;
on a  . Il n'y a donc aucun degré de liberté pour un mélange binaire à l'équilibre liquide-vapeur lorsque la pression et la température sont imposées. À température constante, la pression totale au-dessus du liquide évolue avec la composition de celui-ci comme le montre la figure ci-contre établie avec la loi de Raoult qui donne :
. Il n'y a donc aucun degré de liberté pour un mélange binaire à l'équilibre liquide-vapeur lorsque la pression et la température sont imposées. À température constante, la pression totale au-dessus du liquide évolue avec la composition de celui-ci comme le montre la figure ci-contre établie avec la loi de Raoult qui donne :
- pour le corps :
 ;
;
- pour le corps :
 ;
;
- pour la pression totale :
 ;
;
avec  et
et  les pressions de vapeur saturantes respectives des corps et à la température .
les pressions de vapeur saturantes respectives des corps et à la température .
Néanmoins l'hypothèse de la pression totale constante est prise lors de la démonstration de la relation de Duhem-Margules, ou du moins peut-on démontrer que sa variation est négligeable. Sans cette hypothèse, on obtient :

ou, de façon équivalente :

avec :
- la pression totale ; par définition
 ;
;
 le volume molaire de la phase liquide ;
le volume molaire de la phase liquide ; le volume molaire de la phase vapeur, supposée être un mélange de gaz parfaits :
le volume molaire de la phase vapeur, supposée être un mélange de gaz parfaits :  , avec
, avec  la constante universelle des gaz parfaits.
la constante universelle des gaz parfaits.
Aux points d'extrémum (maximum ou minimum) de pression :

d'où :

De fait, la relation de Duhem-Margules telle qu'établie et énoncée n'est vraie que pour les extrémums de la pression de vapeur, c'est-à-dire pour les azéotropes selon le théorème de Gibbs-Konovalov, et n'est qu'approximative autrement[6].
Démonstration
On suppose un mélange binaire à l'équilibre liquide-vapeur à la pression et la température . Pour la phase liquide, la relation de Gibbs-Duhem à température constante, soit  , donne :
, donne :
- (1)

avec :
- le volume molaire de la phase liquide ;
- la pression ;
- et les fractions molaires respectives des corps et en phase liquide ;
 et
et  les potentiels chimiques respectifs des corps et en phase liquide.
les potentiels chimiques respectifs des corps et en phase liquide.
Puisque le liquide est en équilibre avec sa vapeur, on a l'égalité des potentiels chimiques :


avec  et
et  les potentiels chimiques respectifs des corps et en phase vapeur. Si la phase vapeur peut être considérée comme un mélange de gaz parfaits, alors :
les potentiels chimiques respectifs des corps et en phase vapeur. Si la phase vapeur peut être considérée comme un mélange de gaz parfaits, alors :


avec  et
et  les potentiels chimiques respectifs des corps et en phase vapeur idéale. Si, à température constante, on fait varier la composition de la phase liquide tout en restant à l'équilibre liquide-vapeur, on a :
les potentiels chimiques respectifs des corps et en phase vapeur idéale. Si, à température constante, on fait varier la composition de la phase liquide tout en restant à l'équilibre liquide-vapeur, on a :
- (2a)

- (2b)

La fugacité  d'un corps
d'un corps  quelconque, dans un état quelconque, est définie par la variation à température constante de son potentiel chimique :
quelconque, dans un état quelconque, est définie par la variation à température constante de son potentiel chimique :

Dans un mélange de gaz parfaits la fugacité d'un corps est égale à sa pression partielle  . Ainsi, à température constante :
. Ainsi, à température constante :
- (3)

En considérant les relations (2a), (2b) et (3), on réécrit la relation de Gibbs-Duhem à température constante (1) :


Le volume molaire de la phase gaz idéale valant  , on a :
, on a :
- (4)

Par définition des pressions partielles :

on obtient :
- (5)

En considérant que :
- aux basses pressions, c'est-à-dire loin du point critique du mélange, le volume molaire d'un liquide est très inférieur à celui d'un gaz, soit
 ,
,
- , soit
 et
et  ,
,
on peut écrire, pour des plages de fractions molaires importantes :


Ainsi peut-on réécrire approximativement la relation (5) selon :

- Remarque
- S'il existe en phase gaz un troisième corps, noté
 , incondensable, donc indépendant de la composition du liquide, tel que
, incondensable, donc indépendant de la composition du liquide, tel que  et
et  , alors on a
, alors on a  : on obtient directement la relation ci-dessus à partir de la relation (4). On obtiendrait également plus rapidement cette relation en supposant dès la relation (1) que la pression totale est constante, soit
: on obtient directement la relation ci-dessus à partir de la relation (4). On obtiendrait également plus rapidement cette relation en supposant dès la relation (1) que la pression totale est constante, soit  . Or, comme nous l'avons vu dans l'énoncé de la relation, il est impossible de faire varier la composition de la phase liquide d'un mélange binaire tout en restant à son équilibre liquide-vapeur et à température et pression constantes. Néanmoins, comme démontré ci-dessus, la variation de pression totale engendrée est négligeable devant les variations des pressions partielles qui la composent.
. Or, comme nous l'avons vu dans l'énoncé de la relation, il est impossible de faire varier la composition de la phase liquide d'un mélange binaire tout en restant à son équilibre liquide-vapeur et à température et pression constantes. Néanmoins, comme démontré ci-dessus, la variation de pression totale engendrée est négligeable devant les variations des pressions partielles qui la composent.
En considérant la variable fraction molaire du corps en phase liquide et puisque la démonstration est effectuée à température constante, on a :

Or, puisque  , on a :
, on a :

On obtient finalement la relation de Duhem-Margules :
Relation de Duhem-Margules :
L'expression obtenue sans négliger la variation de pression globale dans la relation (4) est :

ou, à partir de la relation (5) :
Implications
Sens de variation de la pression et des pressions partielles
La relation de Duhem-Margules montre que les pressions partielles de tous les corps évoluent de la même façon en fonction de leur fraction molaire en phase liquide. En effet, selon cette relation  et
et  sont de même signe. Expérimentalement, la pression partielle d'un corps augmente toujours avec sa fraction molaire en phase liquide :
sont de même signe. Expérimentalement, la pression partielle d'un corps augmente toujours avec sa fraction molaire en phase liquide :


La relation de Duhem-Margules est cohérente avec cette observation.
D'autre part, puisque :

bien que la relation de Duhem-Margules soit établie à pression constante, on écrit :

ou :

Ceci permet d'étudier la variation de la pression totale en fonction de la composition du liquide à température constante et d'établir, notamment, les règles de Konovalov.
Règles de Konovalov
La relation de Duhem-Margules permet de démontrer les deux règles de Konovalov, nommées en référence à Dimitri Konovalov qui les établit expérimentalement. En effet :

si et seulement si :

La première règle de Konovalov énonce que, pour un équilibre liquide-vapeur :
« À température donnée, la vapeur est toujours plus riche que le liquide en composant le plus volatil. En d'autres termes, la fraction molaire du composant le plus volatil est toujours plus grande dans la vapeur que dans le liquide. Ou encore, la vapeur est plus riche que le liquide en composant dont l'addition au liquide induit une augmentation de la pression de vapeur totale. »
— Première règle de Konovalov.
L'hypothèse de la pression totale constante prise lors de la démonstration de la relation de Duhem-Margules est donc en contradiction avec cette règle.
La deuxième règle de Konovalov énonce que :
« À température constante, aux points d'extrémum de la pression de vapeur les compositions de la vapeur et du liquide sont identiques. »
— Deuxième règle de Konovalov.
Cette règle décrit donc les azéotropes à température constante. Elle est généralisée par le théorème de Gibbs-Konovalov aux azéotropes à pression constante et aux équilibres de phases autres que l'équilibre liquide-vapeur.
Loi de Raoult et loi de Henry
Si la pression partielle de l'un des composants, par exemple le composant , est proportionnelle à sa fraction molaire en phase liquide (selon la loi de Raoult ou la loi de Henry) :

avec  une constante positive quelconque (qui peut dépendre de la température, mais pas de la composition), alors :
une constante positive quelconque (qui peut dépendre de la température, mais pas de la composition), alors :

Dans ces conditions, la relation de Duhem-Margules induit que :

La pression partielle de l'autre composant, ici le composant , répond donc à la relation :

et par conséquent suit également une relation linéaire avec sa fraction molaire en phase liquide :

avec  une constante positive quelconque (qui peut dépendre de la température, mais pas de la composition).
une constante positive quelconque (qui peut dépendre de la température, mais pas de la composition).
On en déduit que si l'un des composants suit la loi de Raoult ou la loi de Henry, alors l'autre composant suit également l'une de ces deux lois[7].
Correction de Poynting
Si un corps du mélange binaire suit la loi de Raoult ou la loi de Henry avec la correction de Poynting, sa pression partielle est calculée selon :

avec  la fraction molaire du corps en phase liquide et les paramètres qui ne dépendent pas de la composition :
la fraction molaire du corps en phase liquide et les paramètres qui ne dépendent pas de la composition :
 une fonction de la température, ayant la dimension d'une pression:
une fonction de la température, ayant la dimension d'une pression:
- loi de Raoult :
 la pression de vapeur saturante du corps , à la température ;
la pression de vapeur saturante du corps , à la température ;
- loi de Henry :
 la constante de Henry du corps (soluté) dans le deuxième corps
la constante de Henry du corps (soluté) dans le deuxième corps  (solvant) du mélange binaire, à la température , sous la pression
(solvant) du mélange binaire, à la température , sous la pression  ;
;
 un volume molaire de référence :
un volume molaire de référence :
- loi de Raoult :
 le volume molaire du corps pur liquide, à la température , sous la pression ;
le volume molaire du corps pur liquide, à la température , sous la pression ;
- loi de Henry :
 le volume molaire partiel du corps (soluté) infiniment dilué dans le deuxième corps (solvant) du mélange binaire, à la température , sous la pression ;
le volume molaire partiel du corps (soluté) infiniment dilué dans le deuxième corps (solvant) du mélange binaire, à la température , sous la pression ;
- une pression de référence :
- loi de Raoult : la pression de vapeur saturante du corps , à la température ;
- loi de Henry :
 la pression de vapeur saturante du deuxième corps (solvant) du mélange binaire, à la température .
la pression de vapeur saturante du deuxième corps (solvant) du mélange binaire, à la température .
On a alors la dérivée partielle :

Déclinée aux deux corps et d'un mélange binaire, on a :


d'où :

Les lois de Raoult et de Henry supposent que la phase liquide se comporte comme une solution idéale ; son volume molaire vaut par conséquent :

D'autre part, la phase vapeur est supposée être un mélange de gaz parfaits dans la relation de Duhem-Margules comme dans les lois de Raoult et de Henry ; son volume molaire vaut par conséquent :
On obtient :

On retrouve la relation de Duhem-Margules démontrée sans l'hypothèse de la pression constante. On en déduit que si l'un des composants suit la loi de Raoult avec correction de Poynting ou la loi de Henry avec correction de Poynting, alors l'autre composant suit également l'une de ces deux lois[8].
Coefficient d'activité
Margules proposa la solution suivante à la relation[1] - [9] :


avec et les pressions de vapeur saturantes respectives des deux corps à . Il pourrait tout aussi bien s'agir, pour l'un des deux corps, de la constante de Henry. La constante  , déterminée expérimentalement, est identique pour les deux corps. On vérifie la relation de Duhem-Margules :
, déterminée expérimentalement, est identique pour les deux corps. On vérifie la relation de Duhem-Margules :

Selon la valeur de la solution de Margules est capable de représenter des azéotropes, contrairement à la loi de Raoult (qui revient à  ). Cette solution est à l'origine du modèle d'activité de Margules à un paramètre, avec les coefficients d'activité
). Cette solution est à l'origine du modèle d'activité de Margules à un paramètre, avec les coefficients d'activité  des deux corps :
des deux corps :


qui donne, pour les pressions partielles :


Le modèle d'activité de Margules à deux paramètres :
![{\displaystyle \ln \gamma _{\text{A}}={x_{\text{B}}}^{2}\,\left[\alpha +2\left(\beta -\alpha \right)x_{\text{A}}\right]}](https://img.franco.wiki/i/57266768b83760c0947ffcd14d88dd2131cf2dc6.svg)
![{\displaystyle \ln \gamma _{\text{B}}={x_{\text{A}}}^{2}\,\left[\beta +2\left(\alpha -\beta \right)x_{\text{B}}\right]}](https://img.franco.wiki/i/9e98c2c5fcf13b9748d514eade61727cc4c77bb9.svg)
vérifie également la relation de Duhem-Margules :
![{\displaystyle x_{\text{A}}\left({\partial \ln P_{\text{A}} \over \partial x_{\text{A}}}\right)_{T}=x_{\text{B}}\left({\partial \ln P_{\text{B}} \over \partial x_{\text{B}}}\right)_{T}=1-2\,x_{\text{A}}\,x_{\text{B}}\,\left[\alpha \left(3x_{\text{B}}-1\right)+\beta \left(3x_{\text{A}}-1\right)\right]}](https://img.franco.wiki/i/cf744c5355b9f34a3eacbb0ee92267b321bb9b59.svg)
De façon plus générale, la relation de Duhem-Margules impose une contrainte au développement de tout modèle de coefficient d'activité. Un coefficient d'activité dépend de la pression, de la température et de la composition de la phase liquide. Par définition, l'enthalpie libre molaire d'excès  de la phase liquide est égale à :
de la phase liquide est égale à :
Enthalpie libre molaire d'excès : 
et le volume molaire d'excès  est égal à :
est égal à :
Volume molaire d'excès : 
On dérive le coefficient d'activité des deux corps du mélange binaire :


La relation de Gibbs-Duhem à pression et température constantes impose que :

On a par conséquent :

et finalement, avec le volume molaire du gaz parfait :
Relation de Duhem-Margules pour les coefficients d'activité

Le volume molaire idéal  du mélange liquide est égal à :
du mélange liquide est égal à :

avec le volume molaire de référence pour le corps tel que défini au paragraphe Correction de Poynting. Le volume molaire réel du mélange liquide est égal à :

Le volume molaire d'excès est généralement considéré comme négligeable devant le volume molaire idéal : en valeur absolue  . Par exemple, si l'on mélange 1 litre d'eau avec 1 litre d'éthanol, le volume idéal de la solution
. Par exemple, si l'on mélange 1 litre d'eau avec 1 litre d'éthanol, le volume idéal de la solution  est de 2 litres. On obtient cependant un volume réel
est de 2 litres. On obtient cependant un volume réel  d'environ 1,92 litre[10]. Le volume se contracte sous l'effet des forces entre molécules d'eau et d'éthanol : le volume d'excès
d'environ 1,92 litre[10]. Le volume se contracte sous l'effet des forces entre molécules d'eau et d'éthanol : le volume d'excès  est de −0,08 litre. La plupart des modèles de coefficient d'activité (Margules, Van Laar (en), Wilson, NRTL (en), UNIQUAC, UNIFAC, COSMOSPACE) ne dépendent pas de la pression : il est considéré que
est de −0,08 litre. La plupart des modèles de coefficient d'activité (Margules, Van Laar (en), Wilson, NRTL (en), UNIQUAC, UNIFAC, COSMOSPACE) ne dépendent pas de la pression : il est considéré que  et
et  .
.
Expression générale des pressions partielles
En considérant la relation de Duhem-Margules dans sa forme rigoureuse :
les pressions partielles  doivent avoir la forme générale :
doivent avoir la forme générale :

avec pour tout corps :
- la fraction molaire en phase liquide ;
 un coefficient d'activité, fonction de la pression, de la température et de la composition de la phase liquide, sans dimension ;
un coefficient d'activité, fonction de la pression, de la température et de la composition de la phase liquide, sans dimension ;- une fonction de la température, ayant la dimension d'une pression ;
- un volume molaire de référence, fonction de la pression et de la température ;
- une pression de référence, fonction de la température.
La loi de Raoult et la loi de Henry sont retrouvées lorsque le coefficient d'activité est égal à 1 (solution liquide idéale) et la correction de Poynting négligée (aux basses pressions).
Le volume molaire du mélange liquide est égal à :

D'autre part, la relation de Duhem-Margules est développée en considérant que la phase gaz répond à la loi des gaz parfaits, son volume molaire étant  . La relation de Duhem-Margules ne permet donc de trouver des expressions des pressions partielles que pour des pressions de moins de 10 atm. Les modèles de coefficient d'activité ne sont en général développés et valides que dans ce domaine de pression.
. La relation de Duhem-Margules ne permet donc de trouver des expressions des pressions partielles que pour des pressions de moins de 10 atm. Les modèles de coefficient d'activité ne sont en général développés et valides que dans ce domaine de pression.