Microbiome
Le microbiome (du grec micro, « petit », et bios, « vie ») est l'« aire biotique » (aire de vie correspondant à une niche écologique) du microbiote, le mot microbiote désignant ici les espèces autrefois regroupées sous le terme « microflore », c'est-à-dire celles qui prédominent ou sont durablement adaptées à la surface et à l'intérieur d'un organisme vivant[1].
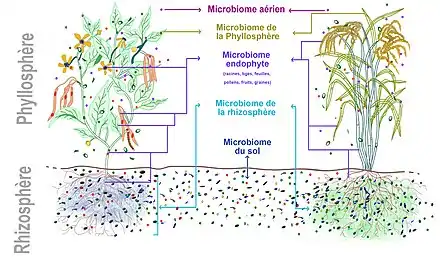
On retrouve aussi sur (voire dans) la plante des microbes plus ou moins ubiquistes et opportunistes, éventuellement pathogènes provenant de l'air et du sol.
L'une de ces espèces (Helicobacter pylori) joue un rôle dans l'ulcère gastro-duodénal.
Ce terme est introduit en 2001 par le généticien et microbiologiste américain Joshua Lederberg pour intégrer la notion d'une communauté écologique comprenant symbiotes, commensaux et pathogènes partageant l'espace corporel humain, dans le but de reconnaître leur fonction de déterminants de la santé et de la maladie[2].
En anglais, le terme microbiome fait référence aux génomes (données génétiques) d'un microbiote. Cette définition ne semble cependant pas faire consensus parmi les auteurs français : d'après Pascale Cossart « on parle de "microbiote" pour désigner l'ensemble des espèces microbiennes présentes dans un environnement, et de "microbiome" quand il s'agit de l'ensemble des gènes présents dans ce microbiote »[3].
Concepts scientifiques mobilisés
Le microbiome est l'expression des conditions écologiques de ces milieux (température, pH, teneurs hormonales, en graisses, en protéines, exposition aux UV, absence de lumière, type de muqueuse, etc.), conditions auxquelles vont répondre les communautés microbiennes en cause, individuellement et/ou collectivement, et qu'elles peuvent modifier ou entretenir à court et moyens termes, mais aussi sur le long terme, c'est-à-dire celui de l'évolution, ou plus précisément de la coévolution du microbiote avec ses hôtes.
Ce concept embrasse les notions de communauté microbienne, de biodiversité microbienne (en nombre d'individus, les microbes sont les organismes les plus nombreux sur terre[4]), d'écologie microbienne et d'interactions durables et fonctionnelles entre micro-organismes, entre eux et l'organisme, ou entre eux et différents organes (allant du simple commensalisme jusqu'à la symbiose, endo- ou ectosymbiose).
Il a, de plus en plus, aussi des bases génétiques. Par extension, le microbiome peut également désigner la somme des génomes des micro-organismes vivant dans ou sur un organisme animal ou végétal (hors état pathologique). Un séquençage collectif de ces organismes est possible (métagénomique), applicable à un écosystème complet[5].
Cette notion est issue du concept de commensalisme théorisé par Pierre-Joseph van Beneden durant la seconde moitié du XIXe siècle[6].
Le mycobiome (en) est la partie du microbiome qui ne concerne que les microchampignons, on parle par exemple de mycobiome humain[7], de mycobiome intestinal (qui pourrait jouer un rôle dans la maladie de Crohn) ou du mycobiome pulmonaire[8]. Il joue un rôle majeur chez de nombreuses plantes[9].
Connaissance
Microbiotes non-humains
Depuis quelques décennies, des chercheurs étudient avec attention le microbiome d'espèces qui nous sont éloignées et qui présentent des capacités particulières de digestion ou de symbiose avec des écosystèmes microbiens particulier, (termites[10] xylophages, insectes herbivores[11] ou reptiles par exemple[12] ou oiseaux comme l'autruche[13] réputée pour son régime alimentaire éclectique) ou proches (primates[14] - [15] par exemple) et d'animaux de rentes (le microbiote des ruminants en particulier[16], qui joue un rôle important pour leur santé)…
Un microbiote des plantes, notamment associé à la rhizosphère est connu depuis plusieurs décennies (composé de bactéries et de champignons dits mycorhyziens), notamment chez les plantes vasculaires. Et des chercheurs ont récemment montré que certains arbres (salicacées) disposent aussi d'endosymbioses dans leur partie aérienne, avec des bactéries fixatrices d'azote qui les aident efficacement à pousser dans des lieux oligotrophes carencés en azote bioassimilable.
Des études ont récemment montrés qu'un écosystème à haute diversité biologique est plus stable et que la biodiversité végétale augmente la productivité d'un milieu[17] - [18]. À des niveaux trophiques "inférieurs", le microbiome végétal a aussi une influence sur la fitness de leurs plantes hôtes[19] - [20] et une hypothèse est que les microbes associés à leurs hôtes végétaux peuvent même influencer le fonctionnement et l'état de l'écosystème via leur rôle dans le modelage du phénotype étendu de leurs organismes hôtes[21] - [22]. Si le degré d'importance du microbiome végétal pour le fonctionnement de l'écosystème n'a pas encore pu être quantifié, une expérience récente (2017) ayant porté sur le lien entre la biodiversité des arbres et le fonctionnement de l'écosystème boisé a néanmoins fortement plaidé en faveur de l'hypothèse que la diversité des bactéries trouvées sur les feuilles de ces arbres est positivement liée à la productivité de l'écosystème entier (même après pris en compte le rôle de la diversité végétale)[23]. Cette étude a aussi conclu que l'identité des espèces hôtes, leur identité fonctionnelle et leur diversité fonctionnelle étaient les principaux déterminants de la structure et de la diversité des communautés bactériennes foliaires. Les auteurs concluent dans ce cas à une corrélation positive entre la diversité microbienne associée à la plante et la productivité de l'écosystème et ils invitent à prendre en compte ce mécanisme pour améliorer les modèles de relations entre biodiversité et écosystème[23].
Étude du microbiome humain
Le microbiote humain, encore très mal connu, fait l'objet de recherches internationales[24] - [25].
Ce n'est qu'en décembre 2007, qu'aux États-Unis a été lancé[26], par le NIH un vaste projet scientifique dénommé Human Microbiome Project visant à séquencer tous les gènes ou génomes, des micro-organismes vivant normalement chez l'homme, à partir d'échantillons, prélevés dans la bouche, la gorge et le nez, sur la peau, dans le tube digestif, et dans le tractus urogénital féminin ainsi (plus récemment) que dans le tractus urogénital masculin[27].
On a montré que ce microbiome est personnel[28] ; il provient en partie de la mère mais aussi du père[27], même s'il est aussi influencé par l'alimentation[29] - [30] et qu'il se diversifie en vieillissant[31]. Il peut aussi acquérir des gènes de bactéries extérieures, par exemple de bactéries marines, qui ont pu être transférées à des bactéries du microbiome de Japonais et y persister[32], probablement sélectionnés pour leur intérêt pour le microbiome et/ou pour l'hôte.
L'épidémiologiste néerlandaise Elisabeth Bik est considérée comme une pionnière dans le microbiome et, en 2016, elle a reçu le Microbiome Pioneer Award, prix réservé aux scientifiques en pointe dans le domaine du microbiome.
Une base de données conçue pour un accès gratuit et facilitant le travail collaboratif, répertoriant le microbiome oral a été ouverte début 2008 par l'Institut américain de recherche dentaire et crâniofaciale (NIDCR) en partenariat avec des chercheurs d'autres pays. Elle contenait déjà 600 micro-organismes.
Grâce au séquençage de l'ARNr 16S, les chercheurs pourront peu à peu classer ces micro-organismes dans un arbre généalogique et mieux comprendre leur importance, par exemple pour l'expression des caries dentaires ou de divers troubles de la digestion.
Outils d'étude métagénomique et phylogénétique du bactériome
Ces « pipelines de données » visent l'analyse de la composition relative d'un échantillon en bactéries, avec par exemple :
- MG-RAST[33].
Outils généralistes (métagénomiques) de profiling taxonomique
Un autre type de « pipeline de données » vise à étudier tous les taxons (connus) d'un même échantillon.
Outils d'étude du virome
Les rôles bénéfiques et parfois pathogènes du microbiome bactérien sont maintenant reconnus, et de plus en plus étudiés[37] - [38], mais le rôle des virus (virome, qui peut affecter la santé de l'hôte (humain ou animal), mais aussi des bactéries[39] - [40] y compris avec les virus de virus) sont encore très mal connus. D'importants changements du virome sont notés, par exemple lors du syndrome d'immunodéficience acquise et de la maladie inflammatoire chronique de l'intestin[41]. Au sein du microbiote, le virome a des liens probablement encore sous-estimés avec la santé et la maladie, chez l'Homme comme dans le reste de la faune, chez les plantes et dans les relations écosystémiques. Une meilleure connaissance du virome est aussi un enjeu pour la santé animale et vétérinaire, pour l'approche One Health recommandée par l'OMS et l'OIE alors que le risque de pandémie zoonotique croît et que les écosystèmes nécessitent de nouveaux outils pour comprendre et étudier la virosphère[37].
L'identification de virus (connus ou inconnus) dans un échantillon composé de multiples microorganismes est plus difficile que celle des bactéries, car les séquences virales sont extrêmement diverses et divergentes[37], souvent sans similitude nucléotidique avec aucune séquence virale connue ou existante[42] - [43]. La bioinformatique utilise alors des comparaisons d'acides aminés, plus difficiles à établir, fondées sur des méthodes encore émergentes ; la découverte de séquences virales se base sur un long travail d'alignement des séquences avec d'autres séquences virales stockées dans les grandes bibliothèques des bases de données génomiques, car, à la différence des bactéries chez lesquelles l'ARN 16S est toujours présente (dans tous les taxons), les virus n'ont pas un tel gène distinctif qui signerait la présence d'un virus, quel que soit son taxon[37].
Les nouveaux outils de la bioinformatique de profilage taxonomique et métagénomique de la biodiversité virale (pipelines d'annotation de données intégrant des algorithmes de traitement dédiés) appliqués au big data qui émergera de l'étude par Séquençage de l'ADN ou de l'ARN de la virosphère pourront contribuer à l'analyse de la composition des viromes. Il devient progressivement possible d'évaluer les types viraux et leur abondance relative dans les échantillons. Les pipelines de données construits pour l'étude de virus sont par exemple :
- VirusSeeker[44],
- Viral Informatics Resource for Metagenome Exploration (VIROME)[45],
- viGEN[46],
- le Viral MetaGenome Annotation Pipeline (VMGAP)[47],
- MetaVir [48],
- Lazypipe, développé par l'Université d'Helskinki qui sous-traite et automatise la recherche d'homologies de gènes viraux à un serveur distinct, permettant ainsi l'utilisation des toutes dernières séquences virales. Cet outil (qui intègre les contigs viraux) utilise le moteur SANSparallel[49] pour rechercher des homologues d'acides aminés dans la base de données UniProtKB[50] ; SANSparallel étant globalement 100 fois plus rapide que le moteur de recherche blastp (utilisé par la plupart des autres pipelines d'annotation selon Somervuo et Holm, 2015)[51].
Des pipelines et algorithmes d'études génomiques sont aussi dédiés au signalement de nouveaux virus encore inconnus de la science[37], avec par exemple :
Évolution des microbiomes
L'évolution des microbiomes se traduit par l'existence de patrons de phylosymbiose[58], dans lesquels les compositions du microbiote de deux espèces sont d'autant plus similaires que ces espèces sont proches phylogénétiquement. Ce phénomène a été retrouvé pour différents genres d'arthropodes, chez les grands singes[59].
La co-évolution mammifères-microbes constitue un « paradoxe immunologique », appelé aussi « inflammation physiologique ». Ce paradoxe revient à se demander par quels mécanismes les mammifères peuvent en même temps conjuguer une tolérance[60] aux microorganismes symbiotes / commensaux (symbiontes) et une élimination des microorganismes pathogènes (pathobiontes). D'où l'émergence d'un nouveau paradigme scientifique selon lequel le développement du système immunitaire est lié au maintien du microbiome[61].
Les microbes se reproduisent et évoluent à un rythme plus rapide que celui des cellules de leurs hôtes, et ils évoluent ou co-évoluent en fonction de facteurs qui sont encore difficiles à appréhender[4], souvent sous le contrôle de l'hôte[62] et/ou d'équilibres entretenus par la diversité de la flore et certains virus[63], tant qu'il est en bonne santé.
Ils peuvent en outre bénéficier de transferts horizontaux de gènes (entre cellules, mais aussi entre espèces différentes[64] - [65]) notamment dans le microbiome intestinal[66], transferts qui semblent plus rares et difficiles entre les organismes plus complexes, notamment animaux[4].
La connaissance de l'évolution des microbiomes est encore lacunaire. Les connaissances dans ce domaine proviennent souvent de modèles très simplificateurs et d'observations biologiques basées sur des observations limitées de communautés très simplifiées[4].
La science est en train de vérifier si les modèles de sélection et d'évolution des théories écologiques et écosystémiques classiques (issues de l'observation et de l'étude des relations entre macro-organismes et leurs écosystèmes) conviennent aussi, ou pas, aux microbiomes pour lesquels les pressions de sélections semblent s'appliquer différemment ou à des rythmes différents[67] - [4] - [68].
Un des enjeux d'une meilleure compréhension du fonctionnement des microbiomes est la santé, humaine et animale, car les déséquilibres ou "maladies du microbiome" semblent aussi pouvoir affecter la santé des hôtes (par exemple concernant la production endogène par la flore intestinale de certaines vitamines[69], la santé sexuelle - avec le microbiome vaginal[70] ou prépucial et du pénis[71] - ou le risque d'obésité[72] - [73]).
Notes et références
- Stéphane Blanc, Gilles Boëtsch, Martine Hossaert-McKey et François Renaud, Écologie de la santé, Le Cherche midi, (ISBN 9782749154251), p. 184.
- (en) Joshua Lederberg et Alexa T. McCray, « 'Ome sweet 'omics : a genealogical treasury of Words », Scientist, vol. 15, no 7, , p. 8 (lire en ligne, consulté le ).
- Cossart 2016, p. 90.
- Yeoman, C.J.; Chia, N.; Yildirim, S.; Miller, M.E.B.; Kent, A.; Stumpf, R.; Leigh, S.R.; Nelson, K.E.; White, B.A.; Wilson, B.A.(2011) Towards an Evolutionary Model of Animal-Associated Microbiomes ; Entropy 2011, 13, 570-594 (résumé), article publié en licence CC-BY-SA
- « Définition | Microbiome », sur futura-sciences.com (consulté le ).
- Brice Poreau, Biologie et complexité : histoire et modèles du commensalisme (Thèse de doctorat en Histoire, philosophie et épistémologie des sciences, des techniques et des technologies), Lyon 1, (lire en ligne).
- Françoise Botterel, Cécile Angebault et Marie-Elisabeth Bougnoux, « Le mycobiome humain : actualités et perspectives », Revue Francophone des Laboratoires, microbiote humain, vol. 2015, no 469, , p. 67–73 (ISSN 1773-035X, DOI 10.1016/S1773-035X(15)72823-6).
- Linh Nguyen et Laurence Delhaes, « Un nouveau concept - Le mycobiome pulmonaire », médecine/sciences, vol. 31, no 11, , p. 945–947 (ISSN 0767-0974 et 1958-5381, DOI 10.1051/medsci/20153111002, lire en ligne, consulté le ).
- (en) Kunihiko Hata, Kazuyoshi Futai et Mitsuya Tsuda, « Seasonal and needle age-dependent changes of the endophytic mycobiota in Pinus thunbergii and Pinus densiflora needles », Canadian Journal of Botany, vol. 2, no 76, , p. 245-250 (DOI 10.1139/B97-177).
- (en) Falk Warnecke, Peter Luginbühl, Natalia Ivanova et Majid Ghassemian, « Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite », Nature, vol. 450, no 7169, , p. 560–565 (ISSN 1476-4687, PMID 18033299, DOI 10.1038/nature06269, lire en ligne, consulté le ).
- (en) Garret Suen, Jarrod J. Scott, Frank O. Aylward et Sandra M. Adams, « An Insect Herbivore Microbiome with High Plant Biomass-Degrading Capacity », PLoS Genetics, vol. 6, no 9, (PMID 20885794, DOI 10.1371/journal.pgen.1001129, lire en ligne, consulté le ).
- (en) Elizabeth K. Costello, Jeffrey I. Gordon, Stephen M. Secor et Rob Knight, « Postprandial remodeling of the gut microbiota in Burmese pythons », The ISME Journal, vol. 4, no 11, , p. 1375–1385 (ISSN 1751-7370, DOI 10.1038/ismej.2010.71).
- (en) Hiroki Matsui, Yuko Kato, Tohru Chikaraishi et Masanori Moritani, « Microbial diversity in ostrich ceca as revealed by 16S ribosomal RNA gene clone library and detection of novel Fibrobacter species », Anaerobe, vol. 16, no 2, , p. 83–93 (ISSN 1075-9964, DOI 10.1016/j.anaerobe.2009.07.005, lire en ligne, consulté le ).
- (en) Suleyman Yildirim, Carl J. Yeoman, Maksim Sipos et Manolito Torralba, « Characterization of the Fecal Microbiome from Non-Human Wild Primates Reveals Species Specific Microbial Communities », PLoS ONE, vol. 5, no 11, , e13963 (ISSN 1932-6203, DOI 10.1371/journal.pone.0013963, lire en ligne, consulté le ).
- (en) Angel J. Rivera, Jeremy A. Frank, Rebecca Stumpf et Abigail A. Salyers, « Differences between the normal vaginal bacterial community of baboons and that of humans », American Journal of Primatology, vol. 73, no 2, , p. 119–126 (ISSN 0275-2565, DOI 10.1002/ajp.20851, lire en ligne, consulté le ).
- (en) Jennifer M. Brulc, Dionysios A. Antonopoulos, Margret E. Berg Miller et Melissa K. Wilson, « Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases », Proceedings of the National Academy of Sciences, vol. 106, no 6, , p. 1948–1953 (ISSN 0027-8424 et 1091-6490, DOI 10.1073/pnas.0806191105, lire en ligne, consulté le ).
- Tilman, D., Reich, P. B. & Isbell, F. Biodiversity impacts ecosystem productivity as much as resources, disturbance, or herbivory. Proc. Natl Acad. Sci. USA 109, 10394–10397 (2012)
- Liang, J. et al. Positive biodiversity-productivity relationship predominant in global forests. Science 354, aaf8957 (2016)
- Vandenkoornhuyse, P., Quaiser, A., Duhamel, M., Le Van, A. & Dufresne, A. The importance of the microbiome of the plant holobiont. New Phytol. 206, 1196–1206 (2015)
- Vorholt, J. A. Microbial life in the phyllosphere. Nat. Rev. Microbiol. 10, 828–840 (2012)
- Bringel, F. & Couée, I. Pivotal roles of phyllosphere microorganisms at the interface between plant functioning and atmospheric trace gas dynamics. Front. Microbiol. 6, 486 (2015)
- Müller, D. B., Vogel, C., Bai, Y. & Vorholt, J. A. The plant microbiota: systems biology insights and perspectives. Annu. Rev. Genet. 50, 211–234 (2016)
- Isabelle Laforest-Lapointe, Alain Paquette Christian Messier & Steven W. Kembel (2017) Leaf bacterial diversity mediates plant diversity and ecosystem function relationships ; Nature ; doi:10.1038/nature22399, mis en ligne le 24 mai 2017
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I (2007) The human microbiome project. Nature, 449, 804–810
- Human Microbiome Jumpstart Reference Strains Consortium. A catalog of reference genomes from the human microbiome. Science 2010, 328, 994–999.
- « Scientists Launch First Comprehensive Database of Human Oral Microbiome » ; Communiqué de presse du NIH, daté du 25 mars 2008
- Angela B. Javurek, William G. Spollen, Amber M. Mann Ali, Sarah A. Johnson, Dennis B. Lubahn, Nathan J. Bivens, Karen H. Bromert, Mark R. Ellersieck, Scott A. Givan & Cheryl S. Rosenfeld (2016)Discovery of a Novel Seminal Fluid Microbiome and Influence of Estrogen Receptor Alpha Genetic Status; Scientific Reports 14 mars 2016 ; doi:10.1038/srep23027
- Benson, A.K.; Kelly, S.A.; Legge, R.; Ma, F.; Low, S.J.; Kim, J.; Zhang, M.; Oh, P.L.; Nehrenberg, D.; Hua, K.; et al. (2010) Individuality in gut microbiome composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc. Natl. Acad. Sci. USA , 107, 18933–18938
- Turnbaugh, P.J.; Ridaura, V.; Faith, J.J.; Rey, F.; Knight, R.; Gordon, J.I (2009) The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med, 1, 6–14
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. (2010) Impact of diet in shaping gut microbiome revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA, 107, 14691–14696.
- Mihajlovski, A.; Doré, J.; Levenez, F.; Monique, A.; Brugère, J (2010) Molecular evaluation of the human gut methanogenic archaeal microbiome reveals an age-associated increase in diversity. Environ. Microbiol. Rep, 2, 272–280
- Hehemann, J.H.; Correc, G.; Barbeyron, T.; Helbert, W.; Czjzek, M.; Michel, G. (2010) ; Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiome. Nature , 464, 908–912.
- Meyer,F. et al. (2008) The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics, 9, 386.
- Truong,D.T. et al. (2015) MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods, 12, 902.
- Wood,D.E. et al. (2019) Improved metagenomic analysis with Kraken 2. Genome Biol., 20, 25
- Kim,D. et al. (2016a) Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res., 26, 1721–1729.
- (en) Ilya Plyusnin, Ravi Kant, Anne J. Jaaskelainen et Tarja Sironen, « Novel NGS Pipeline for Virus Discovery from a Wide Spectrum of Hosts and Sample Types », BioRxiv, Bioinformatics, (DOI 10.1101/2020.05.07.082107, lire en ligne, consulté le )
- Biedermann,L. and Rogler,G. (2015) The intestinal microbiota: its role in health and disease. Eur. J. Pediatr., 174, 151–167.
- Lim,E.S. et al. (2015) Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat. Med., 21, 1228–1234.
- Neil,J.A. and Cadwell,K. (2018) The Intestinal Virome and Immunity. J. Immunol., 201, 1615–1624
- Norman,J.M. et al. (2015) Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell, 160, 447–460.
- Wang,Q. et al. (2013) VirusFinder: software for efficient and accurate detection of viruses and their integration sites in host genomes through next generation sequencing data. PloS One, 8, e64465
- Takeuchi,F. et al. (2014) MePIC, metagenomic pathogen identification for clinical specimens. Jpn. J. Infect. Dis., 67, 62–65.
- Zhao,G. et al. (2017) VirusSeeker, a computational pipeline for virus discovery and virome composition analysis. Virology, 503, 21–30
- Wommack,K.E. et al. (2012) VIROME: a standard operating procedure for analysis of viral metagenome sequences. Stand. Genomic Sci., 6, 427–439
- Bhuvaneshwar,K. et al. (2018) viGEN: An Open Source Pipeline for the Detection and Quantification of Viral RNA in Human Tumors. Front. Microbiol., 9, 1172
- Lorenzi,H.A. et al. (2011) TheViral MetaGenome Annotation Pipeline(VMGAP):an automated tool for the functional annotation of viral Metagenomic shotgun sequencing data. Stand. Genomic Sci., 4, 418–429
- Roux,S. et al. (2014) Metavir 2: new tools for viral metagenome comparison and assembled virome analysis. BMC Bioinformatics, 15, 76.
- Roux,S. et al. (2014) Metavir 2: new tools for viral metagenome comparison and assembled virome analysis. BMC Bioinformatics, 15, 76.
- Zhao,G. et al. (2017) VirusSeeker, a computational pipeline for virus discovery and virome composition analysis. Virology, 503, 21–30.
- Somervuo,P. and Holm,L. (2015) SANSparallel : interactive homology search against Uniprot. Nucleic Acids Res., 43, W24–W29
- Vilsker,M. et al. (2019) Genome Detective: an automated system for virus identification from high- throughput sequencing data. Bioinforma. Oxf. Engl., 35, 871–873.
- Li,Y. et al. (2016) VIP: an integrated pipeline for metagenomics of virus identification and discovery. Sci. Rep., 6, 23774.
- Kostic,A.D. et al. (2011) PathSeq: software to identify or discover microbes by deep sequencing of human tissue. Nat. Biotechnol., 29, 393–396
- Ho,T. and Tzanetakis,I.E. (2014) Development of a virus detection and discovery pipeline using next generation sequencing. Virology, 471–473, 54–60
- Naeem,R. et al. (2013) READSCAN: a fast and scalable pathogen discovery program with accurate genome relative abundance estimation. Bioinforma. Oxf. Engl., 29, 391–392.
- Fosso,B. et al. (2017) MetaShot: an accurate workflow for taxon classification of host-associated microbiome from shotgun metagenomic data. Bioinformatics, 33, 1730–1732.
- (en) Brucker RM, Bordenstein SR, « The hologenomic basis of speciation: gut bacteria cause hybrid lethality in the genus Nasonia », Science, vol. 341, no 6246, , p. 667-669 (DOI 10.1126/science.1240659).
- Thierry Lefevre, Michel Raymond et Frédéric Thomas, Biologie évolutive, De Boeck Superieur, , p. 625.
- Mécanismes de tolérogénèse passive (furtivité des microbiotes) et active (production d'anticorps, compartimentalisation).
- (en) Eric T. Harvill, « Cultivating our "frienemies": viewing immunity as microbiome management », mBio, vol. 4, no 2, (DOI 10.1128/mBio.00027-13).
- Oh, P.L.; Benson, A.K.; Peterson, D.A.; Patil, P.B.; Moriyama, E.N.; Roos, S.; Walter, J. (2010) Diversification of the gut symbiont Lactobacillus reuteri as a result of host-driven evolution. ISME , 4, 377–387.
- Pal, C.; Maciá, M.D.; Oliver, A.; Schachar, I.; Buckling, A. (2007), Coevolution with viruses drives the evolution of bacterial mutation rates. Nature, 450, 1079–1081
- Garcia-Vallvé, S.; Romeu, A.; Palau, J. (2000) Horizontal gene transfer in bacterial and archaeal complete genomes. Genome Res, 10, 1719
- Cohen, O.; Pupko, T. (2010), Inference and characterization of horizontally transferred gene families using stochastic mapping. Mol. Biol. Evol, 27, 703
- Qu, A.; Brulc, J.M.; Wilson, M.K.; Law, B.F.; Theoret, J.R.; Joens, L.A.; Konkel, M.E.; Angly, F.; Dinsdale, E.A.; Edwards, R.E.; Nelson, K.E.; White, B.A. (2008), Comparative metagenomics reveals host-specific metavirulomes and horizontal gene transfer elements in the chicken cecum microbiome. PLoS ON, 3, e2945
- Prosser, J.I.; Bohannan, B.J.M.; Curtis, T.P.; Ellis, R.J.; Firestone, M.K.; Freckleton, R.P.; Green, J.L.; Green, L.E.; Killham, K.; Lennon, J.J.; et al. (2007), The role of ecological theory in microbial ecology. Nat. Rev. Microbiol, 5, 384–392
- Foster, J.A.; Krone, S.M.; Forney, L.J. (2008), Application of ecological network theory to the human microbiome. Interdiscip. Perspect. Infect. Dis. doi:10.1155/2008/839501
- Hill, M.J. Intestinal flora and endogenous vitamin synthesis. Eur. J. Cancer Prev. 1997, 6 (Suppl. 1), S43–S45.
- Wilson, B.A.; Thomas, S.M.; Ho, M (2010) The human vaginal microbiome. In Metagenomics of The Human Body; Nelson, K.E., Ed.; Springer: New York, NY, USA
- Price, L.B.; Liu, C.M.; Johnson, K.E.; Aziz, M.; Lau, M.K.; Bowers, J.; Ravel, J.; Keim, P.S.; Serwadda, D.; Wawer, M.J.; Gray, R.H (2010) The effects of circumcision on the penis microbiome. PLoS ON, 5, e8422
- Schwiertz, A.; Taras, D.; Schäfer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. (2010) Microbiome and SCFA in lean and overweight healthy subjects. Obesity, 18, 190-195.
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. (2008) A core gut microbiome in obese and lean twins. Nature, 453, 480–484.
Voir aussi
Ouvrages en français
- Pascale Cossart, La nouvelle microbiologie : des microbiotes aux CRISPR, Paris, Éditions Odile Jacob, , 255 p. (ISBN 978-2-7381-3331-1, OCLC 951677729)
- Debré, Patrice, L'Homme microbiotique, Éd. Odile Jacob, 2015, 288 p.
Ouvrages en anglais
- Fraune, S.; Bosch, T.C.G (2007) Long-term maintenance of species-specific bacterial microbiome in the basal metazoan Hydra. Proc. Natl. Acad. Sci. USA, 104, 32.
- Zaura, E.; Keijser, B.J.; Huse, S.M.; Crielaard, W (2009). Defining the healthy “core microbiome” of oral microbial communities. BMC Microbiol, 9, 259
- Vijay-Kumar, M.; Aitken, J.D.; Carvalho, F.A.; Cullender, T.C.; Mwangi, S.; Srinivasan, S.; Sitaraman, S.V.; Knight, R.; Ley, R.E.; Gewirtz, A.T. (2010), Metabolic syndrome and altered gut microbiome in mice lacking toll-like receptor 5. Science, 328, 228–231
- Van Bonn, William et col., Aquarium microbiome response to ninety-percent system water change: Clues to microbiome management, Zoo Biology, 2015, vol. 34, p. 360-367
- Gilbert, JA, Jansson, JK, Knight, R., The Earth Microbiome project: successes and aspirations, BMC Biol. 2014, vol. 12, p. 69.
- Larsen, A.M. et col., Characterization of the gut microbiota of three commercially valuable warmwater fish species, Journal of Applied Microbiology, 2014, vol. 116, p. 1396-1404
- Kramer, A. et al., Maintaining health by balancing microbial exposure and prevention of infection: the hygiene hypothesis versus the hypothesis of early immune challenge, Journal of Hospital Infection, 2013 vol. 83, p. S29-S34.
- Balter, M., Taking Stock of the Human Microbiome and Disease, Science, 2012 vol.336, p. 1246-1247.
- Caporaso, JG, Lauber, CL, Walters, WA, et col., Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms, ISME J. 2012, vol. 6, p. 1621-1624.
- Mitchell, L. et col., Microbial diversity in the deep sea and the underexplored "rare biosphere", Proceedings of the National Academy of Sciences, U S A., 2006, vol. 103(32), pp. 12115-20.
- McFall-Ngai, Margaret J., Unseen forces: the influence of bacteria on animal development, Developmental Biology, 2002 1 fév., vol.242(1), p. 1-14.
Articles connexes
Liens externes
- Notice dans un dictionnaire ou une encyclopédie généraliste :
- (en) Base de données Human Oral Microbiome
- (fr) Le microbiote