Maladie de Vaquez
La maladie de Vaquez ou polyglobulie essentielle ou polycythemia vera ou rubra est un syndrome myéloprolifératif, caractérisé par une polyglobulie (augmentation importante de l'hémoglobine) avec une augmentation de l'hématocrite, du nombre de globules rouges et une augmentation du volume globulaire total.
| Médicament | Uramustine, busulfan, pipobroman, Anagrélide, hydroxyurée, ruxolitinib, peginterféron alfa-2a et pipobroman |
|---|---|
| Spécialité | Hématologie |
| CIM-10 | D45 |
|---|---|
| CIM-9 | 238.4 |
| ICD-O | M9950/3 |
| OMIM | 263300 |
| MedlinePlus | 000589 |
| MeSH | D011087 |
![]() Mise en garde médicale
Mise en garde médicale
Épidémiologie
La maladie de Vaquez est une maladie rare. Son incidence est de 2,8 pour 100 000 habitants, montrant une légère prédilection pour le sexe masculin et se manifestant chez l'adulte d'âge moyen. On a signalé des cas familiaux, mais ils sont rares.
Pathogénie
Il s'agit en fait d'une hyperplasie du tissu hématopoïétique avec prolifération myéloïde clonale prédominant sur la lignée érythrocytaire au niveau médullaire et splénique : la moelle rouge remplit même la diaphyse des os longs et il peut (exceptionnellement) exister des foyers d'hématopoièse extramédullaire. Les 3 lignées médullaires : l'érythroïde, la granuleuse et la mégacaryocytaire participent à cette hyperactivité. Ce fait distingue, entre autres, la polyglobulie essentielle des polyglobulies secondaires.
Une mutation de la Janus kinase 2 (ou JAK2, qui fait partie des tyrosine kinases plasmatique) est retrouvée de manière quasi constante dans les cellules nucléées sanguines. Cette mutation causerait une hypersensibilité à certains facteurs de croissance et une indépendance vis a vis de l'érythropoïétine (EPO). Elle n'est pas cependant spécifique de la maladie de Vaquez et est retrouvée dans d'autres syndromes myéloprolifératifs, et très rarement, chez des sujets normaux[1].
La pathogénie des symptômes repose sur l'accroissement du volume et de la viscosité du sang, qui entraîne comme conséquence :
- un ralentissement de la circulation ;
- une augmentation du travail du cœur ;
- une congestion de la rate.
Diagnostic
Signes cliniques
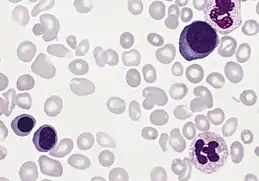
Il s'agit d'une maladie de l'adulte. Âge médian au diagnostic : 60 ans, 5 à 10% < 40 ans.
Elle peut se manifester par :
- une hypertension artérielle ;
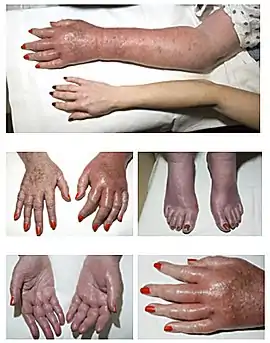
- un prurit et érythrose cutanée (aspect rougeâtre de la peau) ;
- une splénomégalie (rate augmentée de volume).
Elle peut être découverte à l'occasion d'une complication :
- des thromboses veineuses ou artérielles parfois compliquées d'hémorragie ;
- un ulcère gastrique ;
- un priapisme ;
- un syndrome de Budd-Chiari.
Biologie
Le nombre de globules rouges est fortement augmenté, se situant généralement entre 7 et 10 millions par millimètre cube mais peut atteindre 15 millions par mm³. Le taux d'hémoglobine (Hb) et l'hématocrite sont généralement élevés, mais parfois en proportion un peu moindre que le nombre de globules rouges, de sorte que la polyglobulie est alors microcytaire et hypochrome. L'augmentation considérable de l'hématocrite entraîne un accroissement de la viscosité sanguine : quelquefois on éprouve même des difficultés à prendre du sang à la veine. La polyglobulie entraîne également un ralentissement de la VS (vitesse de sédimentation) qui peut même être nulle.
La morphologie globulaire est généralement normale. On peut parfois trouver quelques érythroblastes, notamment après une hémorragie. Plus le nombre d'érythroblastes est élevé, plus le pronostic est sombre. Le taux des réticulocytes est généralement normal sauf après une hémorragie.
Il existe une neutrophilie de l'ordre de 15 000-30 000 polynucléaires neutrophiles/ mm³. Une myélémie peut également être observée.
Le nombre de plaquettes (thrombocytes) peut être augmenté.
Le myélogramme et la biopsie ostéo-médullaire montrent une moelle de couleur rouge foncé et très cellulaire, avec une hyperplasie du tissu myéloïde.
Le fer sérique est normal ou diminué et sa captation par la moelle est rapide. La bilirubinémie est à la limite supérieure de la normale et l'excrétion fécale et urinaire d'urobiline peut être augmentée. Le taux d'acide urique dans le sang est légèrement élevé. On note une excrétion urinaire accrue d'urates et 5 % des sujets présentent des symptômes de lithiase urique ou de goutte.
Le volume sanguin total, déterminé par le marquage des globules rouges par un isotope, est considérablement accru tandis que le volume plasmatique est en principe normal. Cela permet de différencier une vraie polyglobulie d'une hémoconcentration. L'analyse des gaz du sang est utile pour différencier la maladie de Vaquez des polyglobulies d'origine cardio-pulmonaire.
Critères diagnostiques selon l'OMS
L'Organisation mondiale de la santé (OMS) a modifié en 2016 les critères pour le diagnostic de la maladie de Vaquez.
Pour poser le diagnostic, il est nécessaire d'avoir soit trois critères majeurs, soit deux critères majeurs et le critère mineur en l'absence de mutation de JAK2.
Critères majeurs :
- Taux d'hémoglobine supérieur à 16,5 g/dL chez l'homme ou 16 g/dL chez la femme
ou hématocrite supérieur à 49 % chez l'homme ou 48 % chez la femme
ou augmentation du volume globulaire de plus de 25 % par rapport à la normale
- Mutation JAK2 V617F
- Biopsie médullaire montrant une hypercellularité touchant les 3 lignées (panmyélose) avec prolifération mégacaryocytaire
Critères mineurs :
- Taux sériques d'érythropoïétine (EPO) bas
Évolution
La maladie évolue lentement et peut, pendant des décennies, rester semblable à elle-même cependant des complications peuvent assombrir le pronostic. Survie médiane de 14 ans, 24 si patient < 60 ans[2] - [3] - [4].
Complications
- Hémorragies : dues à la polyglobulie (risque au-delà de 55 % d'hématocrite), elles sont fréquentes et parfois dramatiques, elles consistent surtout en épistaxis, hémorragies digestives, rétiniennes (cécité ou restriction variable du champ visuel), cérébrales (généralement fatales) et post-opératoires. Des ecchymoses étendues surviennent au moindre traumatisme, mais le purpura est rare. On observe des hémorragies malgré le nombre élevé de plaquettes car, en cas de caillot, il y a consommation de tous les facteurs de coagulation.
- Thromboses : dues à la thrombopathie, elles sont fréquentes également, elles donnent surtout lieu à des accidents vasculaires cérébraux, tantôt légers (troubles de la parole, parésies), tantôt sévères (paralysies, convulsions) et souvent mortels. Mécanisme des thromboses : voir hémorragies (consommation des facteurs de coagulation).
- La décompensation cardiaque s'installe après plusieurs dizaines d'années d'évolution et est secondaire à l'augmentation du travail cardiaque due à l'hyperviscosité sanguine. À ce moment on va baisser le taux de saturation en oxygène du sang artériel, ce qui peut soulever des difficultés diagnostiques.
- Des complications digestives : l'ulcère gastro-duodénal (généralement duodénal) existe dans 8 % des cas ; une association de la maladie de Vaquez et de cirrhose a été décrite sous le nom de syndrome de Mosse ; des hémorragies digestives et des thromboses des vaisseaux mésentériques et de la veine porte sont des complications assez fréquentes.
- La transformation en une autre maladie myéloproliférative est possible : transformation en myélofibrose avec une énorme rate et une hématopoïèse extramédullaire dans 30 % dans cas après 10-15 ans d'évolution. D'autres (<10 %) se muent en leucémie myéloïde ; on observe alors un stade de transition, dit "érythroleucémique", durant lequel la formule sanguine montre côte-à-côte la polyglobulie et la myélose chronique. La terminaison en leucémie aigüe existe également, de même que la transformation en thrombocytémie (leucémie à plaquettes).
Traitement
Traitement symptomatique
Les complications de la thrombocytose peuvent être prévenues par l'administration d'aspirine à dose antiagrégante (75 à 160 mg/j)
L'hyperuricémie répond à l'allopurinol et l'hydratation alcaline.
Le prurit peut être calmé par des antihistaminique H1 type cétirizine
Traitement spécifique
Il repose sur des saignées. Elles ont l'avantage de soulager rapidement le malade, mais l'inconvénient de déclencher facilement des thromboses suivies parfois d'hémorragies graves ou d'accidents vasculaires cérébraux mortels. Elles sont indiquées pour des sujets jeunes ou lors de grossesse ou en urgence si l'hématocrite est supérieure à 55 %. Elles sont contre-indiquées si les plaquettes sont supérieures à 800 000 par millimètre cube sauf en urgence. Elles entraînent, à la longue, une carence en fer, qui doit être respectée. Il s'agit d'un traitement contraignant avec risque de thrombocytoses secondaires. Les saignées consistent en des prélèvements de 200 à 300 mL avec ou non réinjection du plasma, faits deux à trois fois par semaine en urgence, puis tous les un à trois mois. Le niveau optimal de l'hématocrite à atteindre reste discuté : il semble que viser un niveau plus bas (45 %) pourrait diminuer le risque de complications cardio-vasculaires[5].
L'hydroxyurée ou le pipobroman peut être proposé en cas de sujet non compliants aux saignées ou en cas de thrombocytose. Ces médicaments peuvent être donnés seuls ou en association aux saignées.
Le traitement par le phosphore 32, radioactif, est administré en une fois, par voie intraveineuse, sous la forme de phosphate de sodium marqué au P 32, à la dose de 5 (parfois 10) millicuries. Ce traitement est proposé aux sujets âgés de plus de 75 ans ou avec une espérance de vie limitée. Il est cependant de plus en plus abandonné devant le risque accru de leucémie aigüe. Il est basé sur une injection intra-veineuse unique, suivie d'une deuxième injection après 2 mois en cas d'échec. L'efficacité est retardée de deux mois et nécessite un contrôle la maladie pendant plusieurs années. Il peut être renouvelé en cas de récidive.
Un traitement par interféron peut être proposé chez les patients de moins de 50 ans, sans antécédent thrombotique.
Les biothérapies pourraient modifier la prise en charge future, un inhibiteur de tyrosine kinase, le ruxolitinib, est déjà disponible[6].
La maladie nécessite une surveillance clinique et biologique régulière. Elle est prise en charge, en France, dans le cadre des affections de longue durée (prise en charge à 100 %).
Articles connexes
- Syndrome myéloprolifératif
- Hématologie
- Érythrocytose
Notes et références
- (en)JAK2 Mutations in Polycythemia Vera — Molecular mechanisms and clinical applications, Ayalew Tefferi, New Eng J Med, 2007;356:444-445
- « Polyglobulie primitive (maladie de Vaquez) et thrombocytémie essentielle : mise à jour 2015 », sur www.pjh.fr (consulté le )
- (en) S. Cerquozzi et A. Tefferi, « Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: a literature review of incidence and risk factors », Blood Cancer Journal, vol. 5, , e366 (PMID 26565403, PMCID 4670948, DOI 10.1038/bcj.2015.95, lire en ligne, consulté le )
- (en) A. Tefferi, E. Rumi, G. Finazzi et H. Gisslinger, « Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study », Leukemia, vol. 27, , p. 1874-1881 (ISSN 0887-6924, PMID 23739289, PMCID 3768558, DOI 10.1038/leu.2013.163, lire en ligne, consulté le )
- Marchioli R, Finazzi G, Specchia G et al. Cardiovascular events and intensity of treatment in polycythemia vera, N Engl J Med, 2013;368:22-33
- « John Libbey Eurotext - Hématologie - Place du ruxolitinib dans la polyglobulie de Vaquez, résultats de l’étude internationale d’enregistrement (RESPONSE) : bientôt une autorisation de mise sur le marché en France ! », sur www.jle.com (consulté le )