Déficit en acyl-coenzyme A déshydrogénase des acides gras à chaîne moyenne
Le déficit en acyl-coenzyme A déshydrogénase des acides gras à chaîne moyenne (en anglais Medium-chain acyl-coenzyme A dehydrogenase deficiency), plus connu sous le nom de déficit en MCAD ou MCADD est un trouble de l'oxydation des acides gras qui rend l'organisme incapable de décomposer les chaînes moyennes d'acides gras acyl-coA vers l'acétyl-coA. Ce trouble est responsable d'hypoglycémie pouvant entrainer la mort subite si une intervention rapide de supplémentation en glucose n'est pas engagée ; il survient le plus souvent lors de périodes de jeûne trop importante entre deux repas ou après des vomissements.
| Spécialité | Endocrinologie |
|---|
| CIM-10 | E71.3 |
|---|---|
| CIM-9 | 277.85 |
| OMIM | 201450 |
| DiseasesDB | 7914 |
| eMedicine | 946755 |
| MeSH | C536038 |
![]() Mise en garde médicale
Mise en garde médicale
Avant l'élargissement du dépistage néonatal par le test du buvard, la MCADD était la cause de mort subite du nourrisson la plus fréquemment sous-diagnostiquée. Lorsque la maladie est diagnostiquée avant l'apparition des symptômes, le pronostic des patients est excellent.
La MCADD a longtemps semblé être plus répandue chez les personnes d'Europe du Nord d'origine caucasienne, avec une incidence de 1:4000 à 1:17 000 personnes selon la population. Mais des études ont ensuite remis en cause cette observation.
Le traitement de la MCADD est essentiellement préventif : éviter les situations de jeûne interprandial, c'est-à-dire de trop grand espace de temps entre deux repas et d'autres situations similaires (vomissements, exercice physique prolongé sans apport alimentaire énergétique glucidique supplémentaire...) où l'organisme, qui n'a plus de glucose à disposition, devrait pouvoir compter sur l'oxydation des acides gras pour son approvisionnement en énergie, mais il ne le peut pas, justement en raison de ce déficit enzymatique.
Signes et symptômes
La MCADD se manifeste dans la toute petite enfance par des hypoglycémies hypocétonémiques et un dysfonctionnement hépatique survenant souvent dans des périodes interprandiales trop longues ou à l'occasion d'une infection même mineure entrainant une anorexie et, ou, en présence de vomissements. Les nourrissons qui sont exclusivement nourris au sein peuvent présenter, peu de temps après la naissance, les mêmes symptômes s'ils tètent insuffisamment en quantité ou en fréquence. Chez certains patients, la première manifestation de la MCADD peut être la mort subite, sans cause apparente ou dans le cadre d'une maladie mineure. Un certain nombre de nourrissons porteurs de la MCADD peuvent être complètement asymptomatiques, si les circonstances de ne les ont jamais exposés à une situation d'hypoglycémie. Avec l'avènement de l'élargissement du dépistage néonatal, certaines mères ont rétroactivement été dépistées porteuses de la MCADD, après que leur enfant a été détecté positif au test néonatal du buvard portant sur le niveau de carnitine.
Physiopathologie
Physiologiquement, l'enzyme MCAD est responsable de l'étape de déshydrogénation des acides gras dont la chaîne est longue de 6 à 12 atomes de carbone, lors de la bêta-oxydation à l'intérieur des mitochondries. La bêta-oxydation des acides gras a pour but de fournir de l'énergie à l'organisme lorsque celui-ci a déjà utilisé toutes ses réserves de glucose et de glycogène. Cette oxydation se produit donc pendant les périodes de jeûne, après la période post-prandiale qui, elle, est abondante en glucose, ou lors d'une maladie lorsque l'apport calorique est réduit et souvent en même temps les besoins énergétiques augmentés (c'est le cas par exemple lors d'une infection surtout s'il y a de la fièvre).
Si cette enzyme manque ou ne fonctionne pas, une fois épuisée toute sa réserve de glucose et de glycogène, l'organisme n'a plus aucune source d'énergie, puisqu'il ne peut pas utiliser en dégradant par la déshydrogénase, qui est absente ou défaillante, ses acides gras à chaine moyenne qu'il stocke en particulier dans le foie pour les transformer en glucose.
Génétique
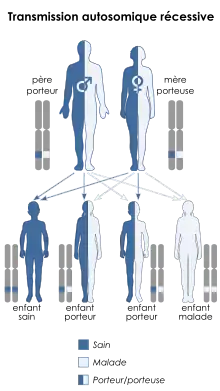
La MCADD est transmise selon le mode autosomique récessif, ce qui signifie qu'une personne touchée a hérité d'un allèle muté (un des deux exemplaires d'un gène) de ses deux parents. Le gène impliqué est appelé ACADM[1], il est situé sur le gène 1, position 31, avec 12 exons et codant une protéine de 421 acides aminés.
Il y a une mutation commune en Europe du Nord, la mutation 985A>G, où une lysine est remplacée par un acide glutamique en position 304 de la protéine. De nombreuses autres mutations ont été identifiées depuis la généralisation du dépistage néonatal, ce qui a élargi le spectre connu des mutations. La mutation 985A>G est présente dans les gènes homozygotes de 80 % des personnes de race blanche qui présentent des signes cliniques de MCADD et chez 60 % des nouveau-nés lorsqu'ils ont été diagnostiqués par le dépistage systématique.
Le génotype d'un individu n'est pas en corrélation directe avec le phénotype en ce qui concerne la MCADD. L'état clinique d'une personne porteuse de la MCADD ne dépend donc pas seulement de la présence de mutations dans son gène ACADM, mais aussi de la présence de stress environnementaux, physiologiques ou pathologiques qui obligent son organisme à dépendre de l'oxydation des acides gras pour produire de l'énergie. Certaines mutations qui ont été identifiées dans les programmes de dépistage néonatal sont associées à une certaine activité enzymatique résiduelle, mais ces types de mutation n'ont pas été observées chez les individus présentant des symptômes cliniques de MCADD. Malgré cela, le traitement préventif par le respect d'un espacement inter-prandial modéré reste la norme pour tous ces individus.
Diagnostic
Cliniquement, la MCADD ou un autre trouble de l'oxydation des acides gras est suspectée chez des personnes qui se présentent avec un état léthargique, des convulsions, un coma ou une hypoglycémie hypocétonémique, en particulier si cet état est déclenché par une maladie mineure. Mais si la MCADD est présente en même temps qu'une maladie hépatique aiguë avec hépatomégalie, cela peut conduire à une confusion diagnostique avec le syndrome de Reye.
Chez certaines personnes, la seule et unique manifestation de la MCADD est la mort subite et inexpliquée, souvent précédée par une maladie mineure qui n'est ordinairement pas létale.
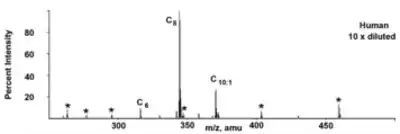
Dans les états où le dépistage des nouveau-nés par la spectrométrie de masse en tandem (MS/MS) est effectué, la MCADD est généralement détectée peu de temps après la naissance, par l'analyse des taches du sang prélevé sur papier buvard. Le profilage de l'acylcarnitine avec la spectrométrie MS/MS dessine une courbe caractéristique de l'élévation de l'hexanoylcarnitine (C6), de l'octanoylcarnitine (C8), de la décanoylcarnitine (C10) ou de la décenoylcarnitine (C10:1), avec la C8 supérieure à la C6 ou la C10.
Une carence en carnitine secondaire est parfois rencontrée chez les patients porteurs de la MCADD, et dans ce cas, le profilage d'acylcarnitine lors de l'analyse en spectrométrie MS/MS peut ne rien montrer de caractéristique. On demande alors une analyse d'urine par chromatographie en phase gazeuse (GC-MS) recherchant la présence d'acide dicarboxylique avec de faibles niveaux de cétone. Sa positivité oriente alors le diagnostic. Des traces d'acylglycine peuvent également être détectées.
Chez les personnes asymptomatiques, les résultats biochimiques des analyses de laboratoire peuvent ne rien révéler. Pour ces personnes, une analyse ciblée d'acylglycine par chromatographie en phase gazeuse (GC-MS), et spécifiquement la recherche et la découverte d'hexanoylglycine et de suberylglycine peuvent aider au diagnostic.
Si les résultats biochimiques orientent fortement vers le diagnostic de MCADD, la génétique moléculaire permettra l'analyse du gène ACADM pour confirmer le diagnostic. L'analyse de l'activité du MCAD dans des cultures de fibroblastes peut également être utilisée pour le diagnostic.
Dans les cas de mort subite hors de tout contexte pathologique ou dans les cas de mort au cours d'une maladie ordinairement bénigne, la MCADD est souvent suspectée. Pour en établir le diagnostic avec certitude, ce qui est très important pour l'information de la famille, l'autopsie montre souvent des dépôts lipidiques dans le foie. La recherche d'acylcarnitine dans la bile et dans le sang peuvent être entreprises pour établir le diagnostic post-mortem. Lorsque les échantillons ne sont pas disponibles, les résidus de sang sur le buvard du dépistage néonatal peuvent être utiles. Des tests biochimiques et génétiques sur la fratrie et les parents peuvent aussi être utiles, même si ceux-ci sont asymptomatiques. La MCADD et les autres désordres de la bêta-oxydation des acides ont été reconnus ces dernières années comme causes non diagnostiquées de la mort subite du nourrisson.
Dépistage
En France
En France, la Haute Autorité de santé recommande le dépistage de la MCADD depuis 2011. Le ministère de la santé a longtemps tardé à équiper les laboratoires chargés des dépistages néonataux pour traiter celui de la MCADD et de plusieurs autres maladies[2] - [3]. Mais depuis le 1er décembre 2020, tous les nouveau-nés sont dépistés pour la MCADD[4].
Ce sont donc désormais 7 maladies, avec la MCADD, qui sont, en France, systématiquement dépistées à la naissance :
la phénylcétonurie depuis 1972, l’hypothyroïdie congénitale depuis 1978, la drépanocytose en Outre-Mer depuis 1985 et 1995 de façon ciblée en métropole, l’hyperplasie congénitale des surrénales depuis 1995, la mucoviscidose depuis 2002 et à la surdité permanente bilatérale depuis 2012[4].
Au Québec
Depuis 2011, les enfants nés au Québec sont dépistés pour la MCADD dès la naissance[5].
Aux États-Unis
À la suite de la recommandation de l'American College of Medical Genetics, les 50 états offrent le dépistage de la MCADD depuis 2006[6].
Traitement
Comme pour la plupart des autres troubles métaboliques liés à l'oxydation des acides gras, les personnes porteuses de la MCADD doivent absolument éviter de jeûner trop longtemps entre deux repas[7]. Ces patients doivent gérer rigoureusement leurs apports alimentaires afin d'éviter toute décompensation métabolique qui est susceptible de leur être fatale. La supplémentation à la demande en glucides simples, tel le glucose, au cours de leur maladie est la clé pour éviter le catabolisme des acides gras que leur maladie empêche de faire correctement et donc leur éviter l'hypoglycémie hypocétonémique.
De 0 à 6 mois
Chez le nourrisson, durant les six premiers mois, le jeûne entre chaque tétée ne doit absolument pas dépasser 3 à 4 heures. L'allaitement maternel reste recommandé, comme pour les nourrissons non atteints de la MCADD. Les substituts de lait maternel enrichis en lipides sont cependant à proscrire.
De 7 mois à 1 an
Durant cette période, les périodes de jeûne entre deux prises alimentaires sont peu à peu allongées, en suivant le principe d'environ une heure de plus par mois :
- 5-6 heures à 7 mois,
- 6-7 heures à 8 mois,
- 7-9 heures à 9 mois,
- 9-10 heures à 10 mois,
- 10-11 heures à 11 mois,
- 11-12 heures à 12 mois.
Mais comme chaque enfant peut réagir différemment à l'espacement des repas, ces directives d'heure des repas peuvent varier d'un patient à l'autre. La règle principale est de ne jamais sauter les repas du matin et du soir (déjeuner ou petit-déjeuner en France et dîner ou souper).
Avec la diversification alimentaire, il est important de respecter désormais certains principes :
- Éviter la charcuterie et viande crue contenant plus de 10 g de gras par 100 g,
- Préférer le lait écrémé et les fromages allégés en gras,
- Manger des fruits et des légumes,
- Le lait de coco, l'huile de palmiste et certains beurres sont très riches en Triglycéride à chaîne moyenne (MCT en anglais), et donc à éviter,
- Les plats doivent contenir des sucres lents,
- Éviter les fritures en alimentation quotidienne.
Après 1 an
Après un an, les règles précédentes s'appliquent. Tout au long de leur vie, les patients atteints de MCADD doivent garder une alimentation saine et pauvre en lipides. Des exceptions à cette bonne diététique sont cependant tolérées de temps en temps.
L'apport énergétique
Durant les premiers mois de vie, le lait maternel et les préparations commerciales couvrent les besoins énergétiques de l'enfant.
Jusqu'à 18 ans, les apports énergétiques issus des lipides recommandés pour les enfants et adolescents sains (sans trouble métabolique) varient de 25 % à 45 % en fonction de l'âge[9]. Pour les patients atteints de MCADD, il est recommandé de ne jamais dépasser 30 %, et ce à n'importe quel âge[8].
Études
Entre 1994 et 2004, une étude a été faite en Australie sur 59 malades atteints de MCADD[10].
Sur ces 59 personnes, 24 ont été diagnostiquées précocement dans leur vie, ont reçu des conseils diététiques et ont été suivies médicalement, tandis que les 35 autres ont été diagnostiquées plus tardivement.
Sur les 24 malades diagnostiqués précocement:
- 3 ont subi une décompensation sévère incluant un décès néonatal,
- 21 ont suivi le traitement diététique et n'ont eu aucun problème.
Sur les 35 malades diagnostiqués tardivement :
- 23 ont subi une décompensation sévère, y compris 5 décès,
- 12 n'ont pas suivi de traitement et n'ont eu aucun problème.
38 des 52 patients vivants ont effectué des tests neurophysiologiques, et cette étude montre que rien ne suggère "des différences significatives dans les résultats cognitifs généraux entre les groupes" de patients diagnostiqués tôt ou tard dans leur vie.
En 2005, une autre étude a été faite aux Pays-Bas[11] sur 155 individus néerlandais atteints de déficience en MCAD, âgés de 3 mois à 5 ans. Elle montre des résultats similaires (mortalité élevée avant le diagnostic, risque d'atteintes neurologiques lors des décompensations), celle-ci montre cependant une prédisposition des personnes atteintes de MCADD à l'obésité et pour un tiers d'entre elles, à des troubles mineurs telles que la fatigue, des douleurs musculaires et une tolérance réduite à l'exercice physique[12].
Incidence
Il a longtemps été écrit que la population caucasienne d'Europe du Nord était la plus exposée à la déficience en MCAD. Mais des études récentes tendent à montrer qu'il s'agit peut-être d'un biais statistique. En effet ce sont les populations chez lesquelles le dépistage est le plus fréquemment fait et les moyens diagnostics les plus disponibles pour les médecins et les plus accessibles financièrement pour les patients. Si on cherche mieux les malades, on en trouve plus. Le corollaire est qu'en cherchant moins bien, on en trouve forcément moins.
Par exemple, le déficit en MCAD a été considéré comme moins courant dans les populations hispaniques, afro-américaines et amérindiennes aux États-Unis. Cependant, cette vision a été corrigée par la mise en œuvre du dépistage systématique chez les nouveau-nés d'origines ethniques différentes. Les données disponibles en Californie démontrent maintenant que la carence en MCAD est aussi répandue chez les Amérindiens (1 : 7 500 naissances) que les natifs d'Europe du Nord, et les prévalences sont similaires chez les nouveau-nés d'origine hispanique, noire et moyen-orientale (1 : 23 000 naissances)[13].
Fondée sur différentes statistiques de détection néonatale du déficit en MCAD, l'incidence suivante a été établie[13] :
| Continent | Pays / État / Région | Incidence |
|---|---|---|
| Asie | 1:51 000 | |
| 1:263 500 | ||
| 1:18 000 | ||
| Océanie | 1:19 000 | |
| Europe | 1:24 900 | |
| 1:9 000 | ||
| 1:10 700 | ||
| 1:4 900 | ||
| 1:8 500 | ||
| 1:13 400 | ||
| 1:8 500 | ||
| 1:16 000 | ||
| 1:23 000 | ||
| 1:12 000 | ||
| 1:8 700 | ||
| Amérique du nord | 1:23 400 | |
| 1:19 000 | ||
| 1:15 000 | ||
| 1:14 000 | ||
| 1:19 000 | ||
| 1:13 000 | ||
| 1:15 700 |
Notes et références
- GeneCards Human Gene Database, « ACADM Gene - GeneCards | ACADM Protein | ACADM Antibody », sur www.genecards.org (consulté le )
- « Dépistage néonatal : la France en retard », Le Figaro, (lire en ligne, consulté le )
- « Vers l'élargissement du dépistage néonatal en France », Le Figaro, (lire en ligne, consulté le )
- Communiqué de presse publié par le ministère de la santé le 30.11.2020 https://solidarites-sante.gouv.fr/actualites/presse/communiques-de-presse/article/le-programme-national-du-depistage-neonatal-evolue
- « Dépistage néonatal sanguin et urinaire : Services Québec – Citoyens », sur www4.gouv.qc.ca (consulté le )
- (en) Claudia Soler-Alfonso, Michael J Bennett et Can Ficicioglu, « Screening for medium-chain acyl CoA dehydrogenase deficiency: current perspectives », Research and Reports in Neonatology, vol. 6, (DOI 10.2147/RRN.S60617, lire en ligne, consulté le )
- (en) « Safe and unsafe duration of fasting for children with MCAD deficiency (PDF Download Available) », sur ResearchGate (consulté le )
- (en) MCADD Guide of Juliane Marie Centre, Denmark (lire en ligne), p. 4
- « Dietary Recommendations for Healthy Children », sur www.heart.org (consulté le )
- Bridget Wilcken, Marion Haas, Pamela Joy et Veronica Wiley, « Outcome of neonatal screening for medium-chain acyl-CoA dehydrogenase deficiency in Australia: a cohort study », Lancet (London, England), vol. 369, no 9555, , p. 37–42 (ISSN 1474-547X, PMID 17208640, DOI 10.1016/S0140-6736(07)60029-4, lire en ligne, consulté le )
- https://www.jpeds.com/article/S0022-3476(05)01189-3/fulltext
- (en) Terry G. J. Derks, Dirk-Jan Reijngoud, Hans R. Waterham et Willem-Jan M. Gerver, « The natural history of medium-chain acyl CoA dehydrogenase deficiency in the Netherlands: Clinical presentation and outcome », The Journal of Pediatrics, vol. 148, no 5, , p. 665–670.e3 (ISSN 0022-3476 et 1090-123X, PMID 16737882, DOI 10.1016/j.jpeds.2005.12.028, lire en ligne, consulté le )
- Dietrich Matern et Piero Rinaldo, GeneReviews(®), University of Washington, Seattle, (PMID 20301597, lire en ligne)