Bioinformatique structurale
La bioinformatique structurale est la branche de la bio-informatique liée à l'analyse et à la prédiction de la structure tridimensionnelle des macromolécules biologiques telles que les protéines, l'ARN et l'ADN. Elle traite des généralisations sur les structures tridimensionnelles des macromolécules, telles que les comparaisons des repliements globaux et des motifs locaux, les principes du repliement moléculaire, l'évolution, les interactions de liaison et les relations structure/fonction, en travaillant à la fois à partir de structures résolues expérimentalement et de modèles informatiques. Le terme structural a la même signification qu'en biologie structurale, et la bio-informatique structurale peut être considérée comme faisant partie de la biologie structurale computationnelle. L'objectif principal de la bio-informatique structurale est la création de nouvelles méthodes d'analyse et de manipulation de données macromoléculaires biologiques afin de résoudre des problèmes de biologie et de générer de nouvelles connaissances[1].
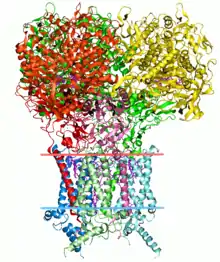
Introduction
Structure des protéines
La structure d'une protéine est directement liée à sa fonction. La présence de certains groupes chimiques à des endroits spécifiques permet aux protéines d'agir comme des enzymes, catalysant plusieurs réactions chimiques[2]. En général, les structures protéiques sont classées en quatre niveaux : primaire (séquences), secondaire (conformation locale de la chaîne polypeptidique), tertiaire (structure tridimensionnelle du pli protéique) et quaternaire (association de plusieurs structures polypeptidiques). La bio-informatique structurale traite principalement des interactions entre les structures en tenant compte de leurs coordonnées spatiales. Ainsi, la structure primaire est mieux analysée dans les branches traditionnelles de la bio-informatique. Cependant, la séquence implique des restrictions qui permettent la formation de conformations locales conservées de la chaîne polypeptidique, telles que l'hélice alpha, les feuillets bêta et les boucles (structure secondaire[3]). De plus, les interactions faibles (telles que les liaisons hydrogène) stabilisent le repliement des protéines. Les interactions peuvent être intra-chaîne, c'est-à-dire lorsqu'elles se produisent entre des parties du même monomère protéique (structure tertiaire), ou inter-chaîne, c'est-à-dire lorsqu'elles se produisent entre différentes structures (structure quaternaire).
Visualisation de la structure

La visualisation de la structure des protéines est un enjeu important pour la bio-informatique structurale[4]. Il permet aux utilisateurs d'observer des représentations statiques ou dynamiques des molécules, permettant également la détection d'interactions qui peuvent être utilisées pour faire des inférences sur les mécanismes moléculaires. Les types de visualisation les plus courants sont :
- Cartoon : ce type de visualisation des protéines met en évidence les différences de structure secondaire. En général, l'hélice α est représentée par un type de vis, les brins β par des flèches et les boucles par des lignes.
- Lignes : chaque résidu d'acide aminé est représenté par des lignes fines, ce qui permet un faible coût de rendu graphique.
- Surface : dans cette visualisation, la forme externe de la molécule est représentée.
- Bâtonnets : chaque liaison covalente entre les atomes d'acides aminés est représentée par un bâtonnet. Ce type de visualisation est le plus utilisé pour visualiser les interactions entre les acides aminés.
Structure de l'ADN
La structure classique des duplex d'ADN a été initialement décrite par Watson et Crick qui se sont appuyés sur les travaux de Rosalind Franklin. La molécule d'ADN est composée de trois substances : un groupe phosphate, un pentose et une base azotée (adénine, thymine, cytosine ou guanine). La structure en double hélice de l'ADN est stabilisée par des liaisons hydrogènes formées entre les paires de bases : l'adénine avec la thymine (AT) et la cytosine avec la guanine (CG). De nombreuses études de bio-informatique structurelle se sont concentrées sur la compréhension des interactions entre l'ADN et les petites molécules, ce qui a été la cible de plusieurs études de conception de médicaments.
Interactions
Les interactions sont des contacts établis entre des parties de molécules à différents niveaux. Ils sont responsables de la stabilisation des structures protéiques et exercent une gamme variée d'activités. En biochimie, les interactions sont caractérisées par la proximité de groupes d'atomes ou de régions de molécules qui présentent un effet les uns sur les autres, tels que les forces électrostatiques, la liaison hydrogène et l'effet hydrophobe. Les protéines peuvent effectuer plusieurs types d'interactions, telles que les interactions protéine-protéine (PPI), les interactions protéine-peptide[5], les interactions protéine-ligand (PLI)[6] et l'interaction protéine-ADN.
.png.webp)
Calculer les contacts
Le calcul des contacts est une tâche importante en bio-informatique structurale, étant important pour la prédiction correcte de la structure et du repliement des protéines, de la stabilité thermodynamique, des interactions protéine-protéine et protéine-ligand, des analyses d'amarrage et de dynamique moléculaire, etc[8].
Traditionnellement, les méthodes de calcul ont utilisé la distance seuil entre les atomes (également appelée cutoff) pour détecter d'éventuelles interactions[9]. Cette détection est effectuée en fonction de la distance euclidienne et des angles entre atomes de types déterminés. Cependant, la plupart des méthodes basées sur la distance euclidienne simple ne peuvent pas détecter les contacts occlus. Par conséquent, les méthodes sans seuil, telles que la triangulation de Delaunay, ont pris de l'importance ces dernières années. De plus, la combinaison d'un ensemble de critères, par exemple, les propriétés physico-chimiques, la distance, la géométrie et les angles, a été utilisée pour améliorer la détermination du contact[8].
| Nature | Critères de distance maximal |
|---|---|
| Liaison hydrogène | 3,9 Å |
| Interaction hydrophobe | 5 Å |
| Interaction ionique | 6 Å |
| Empilage aromatique | 6 Å |
Banque de données sur les protéines (PDB)
.png.webp)
La banque de données sur les protéines (PDB) est une base de données de structure 3D pour les grandes molécules biologiques, telles que les protéines, l'ADN et l'ARN. PDB est géré par une organisation internationale appelée Worldwide Protein Data Bank (wwPDB), qui est composée de plusieurs organisations locales, telles que PDBe, PDBj, RCSB et BMRB. Ils sont responsables de la mise à disposition gratuite de copies des données PDB sur Internet. Le nombre structures disponibles au PDB continue d'augmenter chaque année, étant généralement obtenues par cristallographie aux rayons X, spectroscopie RMN ou cryomicroscopie électronique.
Format des données
Le format PDB (.pdb) est l'ancien format de fichier texte utilisé pour stocker les informations des structures tridimensionnelles des macromolécules utilisées par la banque de données sur les protéines.
En raison de restrictions dans la conception de la structure du format, le format PDB ne permet pas de grandes structures contenant plus de 62 chaînes ou 99999 enregistrements d'atomes[10].
Le PDBx/mmCIF (fichier d'informations cristallographiques macromoléculaires) est un format de fichier texte standard pour représenter les informations cristallographiques[11]. Depuis 2014, le format PDB a été remplacé comme distribution d'archives PDB standard par le format de fichier PDBx/mmCIF (.cif). Alors que le format PDB contient un ensemble d'enregistrements identifiés par un mot-clé de six caractères maximum, le format PDBx/mmCIF utilise une structure basée sur la clé et la valeur, où la clé est un nom qui identifie une caractéristique et la valeur est l'information variable[12].
Autres bases de données structurelles
En plus de la Protein Data Bank (PDB), il existe plusieurs bases de données de structures protéiques et d'autres macromolécules. Les exemples comprennent :
- MMDB : structures tridimensionnelles déterminées expérimentalement de biomolécules issues de la Protein Data Bank (PDB)[13].
- Base de données sur les acides nucléiques (NDB) : informations déterminées expérimentalement sur les acides nucléiques (ADN, ARN)[14].
- Classification structurale des protéines (SCOP) : description complète des relations structurelles et évolutives entre des protéines structurellement connues[15].
- TOPOFIT-DB : alignements structuraux de protéines basés sur la méthode TOPOFIT[16].
- Electron Density Server (EDS) : cartes et statistiques de densité électronique sur l'ajustement des structures cristallines et leurs cartes[17].
- CASP : Prediction Center, expérience communautaire et mondiale de prédiction de la structure des protéines CASP[18].
- Serveur PISCES pour la création de listes de protéines non redondantes : génère une liste PDB par identité de séquence et critères de qualité structurelle[19].
- SBKB (Structural Biology KnowledgeBase) : outils d'aide à la conception de la recherche sur les protéines[20].
- ProtCID (Protein Common Interface Database) : base de données d'interfaces protéine-protéine similaires dans les structures cristallines de protéines homologues[21].
- AlphaFold : base de données sur la structure des protéines[22].
Comparaison de structures
Alignement structurel
L'alignement structurel est une méthode de comparaison entre des structures 3D en fonction de leur forme et de leur conformation[23]. Il pourrait être utilisé pour déduire la relation évolutive entre un ensemble de protéines, même avec une faible similitude de séquence. L'alignement structurel implique la superposition d'une structure 3D sur une seconde, la rotation et la translation des atomes dans des positions correspondantes (en général, en utilisant les atomes C α ou même les atomes lourds du squelette C, N, O et C α). Habituellement, la qualité de l'alignement est évaluée sur la base de l'écart quadratique moyen (RMSD) des positions atomiques, c'est-à-dire la distance moyenne entre les atomes après superposition :

où δ i est la distance entre l'atome i et soit un atome de référence correspondant dans l'autre structure, soit la coordonnée moyenne des N atomes équivalents. En général, le résultat RMSD est mesuré en unité Ångström (Å), ce qui équivaut à 10 −10 m. Plus la valeur RMSD est proche de zéro, plus les structures sont similaires.
Signatures structurelles basées sur des graphes
Les signatures structurelles, également appelées empreintes digitales, sont des représentations de modèles de macromolécules qui peuvent être utilisées pour déduire des similitudes et des différences. Les comparaisons entre un grand nombre de protéines à l'aide de RMSD constituent toujours un défi en raison du coût de calcul élevé des alignements structurels. Des signatures structurelles basées sur des modèles de distance de graphe entre des paires d'atomes ont été utilisées pour déterminer des vecteurs d'identification de protéines et pour détecter des informations non triviales[24]. De plus, l'algèbre linéaire et l'apprentissage automatique peuvent être utilisés pour regrouper les signatures protéiques, détecter les interactions protéine-ligand, prédire ΔΔG et proposer des mutations basées sur la distance euclidienne[25].
Prédiction de structure
Les structures atomiques des molécules peuvent être obtenues par plusieurs méthodes, telles que la cristallographie aux rayons X (XRC), la spectroscopie RMN et la microscopie électronique 3D ; cependant, ces processus peuvent présenter des coûts élevés et parfois certaines structures peuvent être difficilement établies, telles que les protéines membranaires. Par conséquent, il est nécessaire d'utiliser des approches informatiques pour déterminer les structures 3D des macromolécules. Les méthodes de prédiction de structure sont classées en modélisation comparative et modélisation de novo.
Modélisation comparative
La modélisation comparative, également appelée modélisation par homologie, correspond à la méthodologie pour construire des structures tridimensionnelles à partir d'une séquence d'acides aminés d'une protéine cible et d'une matrice de structure connue. La littérature a décrit que les protéines liées à l'évolution ont tendance à présenter une structure tridimensionnelle conservée[26]. En outre, des séquences de protéines apparentées distantes dont l'identité est inférieure à 20 % peuvent présenter des replis différents[27].
Modélisation de novo
En bioinformatique structurale, la modélisation ab initio, également connue sous le nom de modélisation ab initio, fait référence à des approches permettant d'obtenir des structures tridimensionnelles à partir de séquences sans la nécessité d'une structure 3D connue homologue. Malgré les nouveaux algorithmes et méthodes proposés ces dernières années, la prédiction de la structure des protéines de novo est toujours considérée comme l'un des problèmes restants en suspens dans la science moderne[28].
Validation des structures
Après la modélisation de la structure, une étape supplémentaire de validation de la structure est nécessaire car de nombreux algorithmes et outils de modélisation comparative et « de novo » utilisent des heuristiques pour essayer d'assembler la structure 3D, ce qui peut générer de nombreuses erreurs. Certaines stratégies de validation consistent à calculer des scores énergétiques et à les comparer à des structures déterminées expérimentalement. Par exemple, le score DOPE est un score énergétique utilisé par l' outil MODELLER pour déterminer le meilleur modèle[29].
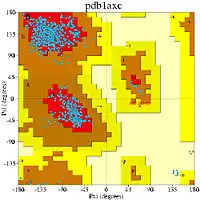
Une autre stratégie de validation consiste à calculer les angles dièdres du squelette φ et ψ de tous les résidus et à construire un diagramme de Ramachandran. La chaîne latérale des acides aminés et la nature des interactions dans le squelette restreignent ces deux angles, et ainsi, la visualisation des conformations autorisées pourrait être effectuée sur la base du graphique de Ramachandran. Une grande quantité d'acides aminés alloués dans des positions non permissives du graphique est une indication d'une modélisation de mauvaise qualité.
Outils de prédiction
Une liste des outils logiciels couramment utilisés pour la prédiction de la structure des protéines, y compris la modélisation comparative, le filetage des protéines, la prédiction de la structure des protéines de novo et la prédiction de la structure secondaire est disponible dans la liste des logiciels de prédiction de la structure des protéines.
Amarrage moléculaire

L'amarrage moléculaire (également appelé docking) est une méthode utilisée pour prédire les coordonnées d'orientation d'une molécule (ligand) lorsqu'elle est liée à une autre (récepteur ou cible). La liaison peut se faire principalement par le biais d'interactions non covalentes, tandis que la liaison liée de manière covalente peut également être étudiée. L'amarrage moléculaire vise à prédire les poses possibles (modes de liaison) du ligand lorsqu'il interagit avec des régions spécifiques du récepteur. Les outils d'amarrage utilisent des champs de force pour estimer un score permettant de classer les meilleures poses qui ont favorisé de meilleures interactions entre les deux molécules.
En général, les protocoles d'amarrage sont utilisés pour prédire les interactions entre les petites molécules et les protéines. Cependant, l'amarrage peut également être utilisé pour détecter des associations et des modes de liaison entre des protéines, des peptides, des molécules d'ADN ou d'ARN, des glucides et d'autres macromolécules.
Dépistage virtuel
Le criblage virtuel (VS) est une approche informatique utilisée pour le criblage rapide de grandes bibliothèques de composés pour la découverte de médicaments. Habituellement, le criblage virtuel utilise des algorithmes d'amarrage pour classer les petites molécules ayant la plus haute affinité pour un récepteur cible.
Ces derniers temps, plusieurs outils ont été utilisés pour évaluer l'utilisation du criblage virtuel dans le processus de découverte de nouveaux médicaments. Cependant, des problèmes tels que des informations manquantes, une compréhension inexacte des propriétés moléculaires de type médicament, des fonctions de notation faibles ou des stratégies d'amarrage insuffisantes entravent le processus d'amarrage. Par conséquent, la littérature a décrit qu'elle n'est toujours pas considérée comme une technologie mature[30] - [31].
Dynamique moléculaire
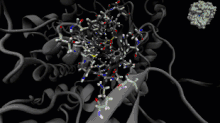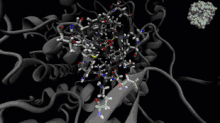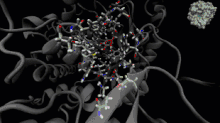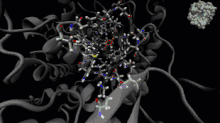
La dynamique moléculaire est une méthode de calcul pour simuler les interactions entre les molécules et leurs atomes pendant une période de temps donnée[33]. Cette méthode permet l'observation du comportement des molécules et de leurs interactions, en considérant le système dans son ensemble. Pour calculer le comportement des systèmes et, ainsi, déterminer les trajectoires, la dynamique moléculaire peut utiliser l'équation du mouvement de Newton, en plus d'utiliser des méthodes de mécanique moléculaire pour estimer les forces qui se produisent entre les particules (champs de force)[34].
Applications
Les approches informatiques utilisées en bioinformatique structurale sont :
- Sélection de la cible - Les cibles potentielles sont identifiées en les comparant avec des bases de données de structures et de séquences connues. L'importance d'une cible peut être décidée sur la base de la littérature publiée. La cible peut également être sélectionnée sur la base de son domaine protéique. Les domaines protéiques sont des blocs de construction qui peuvent être réarrangés pour former de nouvelles protéines. Ils peuvent être étudiés isolément dans un premier temps.
- Suivi des essais de cristallographie aux rayons X - La cristallographie aux rayons X peut être utilisée pour révéler la structure tridimensionnelle d'une protéine. Mais, afin d'utiliser les rayons X pour étudier les cristaux de protéines, des cristaux de protéines pures doivent être formés, ce qui peut prendre de nombreux essais. Cela conduit à un besoin de suivre les conditions et les résultats des essais. De plus, des algorithmes d'apprentissage automatique supervisé peuvent être utilisés sur les données stockées pour identifier les conditions susceptibles d'augmenter le rendement en cristaux purs.
- Analyse des données cristallographiques des rayons X - Le diagramme de diffraction obtenu à la suite du bombardement des rayons X sur les électrons est la transformée de Fourier de la distribution de la densité électronique. Il existe un besoin d'algorithmes capables de déconvoluer la transformée de Fourier avec des informations partielles (en raison d'informations de phase manquantes, car les détecteurs ne peuvent mesurer que l'amplitude des rayons X diffractés, et non les déphasages). Une technique d'extrapolation telle que la dispersion anormale à plusieurs longueurs d'onde peut être utilisée pour générer une carte de densité électronique, qui utilise l'emplacement des atomes de sélénium comme référence pour déterminer le reste de la structure. Le modèle boule-et-bâton standard est généré à partir de la carte de densité électronique.
- Analyse des données de spectroscopie RMN - Les expériences de spectroscopie par résonance magnétique nucléaire produisent des données bidimensionnelles (ou plus), chaque pic correspondant à un groupe chimique dans l'échantillon. Des méthodes d'optimisation sont utilisées pour convertir les spectres en structures tridimensionnelles.
- Corréler les informations structurelles avec les informations fonctionnelles - Les études structurelles peuvent être utilisées comme sonde pour la relation structurelle-fonctionnelle.
Outils
| Logiciel | La description |
|---|---|
| I-TASSER | Prédiction d'un modèle de structure tridimensionnelle de molécules de protéines à partir de séquences d'acides aminés. |
| MEO | Molecular Operating Environment (MOE) est une plate-forme étendue comprenant la modélisation structurelle des protéines, des familles de protéines et des anticorps[35]. |
| SBL | La bibliothèque de bioinformatique structurale : applications pour utilisateurs finaux et algorithmes avancés |
| BALLVoir | Modélisation moléculaire et visualisation[36] |
| PIQÛRE | Visualisation et analyse |
| PyMOL | Visionneuse et modélisation[37] |
| VMD | Visionneuse, dynamique moléculaire[38] |
| Roi | Une visionneuse de kinemage Java open source |
| FOULÉE | Détermination de la structure secondaire à partir des coordonnées[39] |
| MolProbité | Serveur web de validation de structure |
| PROCHECK | Un web service de validation de structure |
| CheShift | Une application en ligne de validation de la structure des protéines |
| 3D-mol.js | Un visualiseur moléculaire pour les applications Web développées en Javascript |
| PROPKA | Prédiction rapide des valeurs de pKa des protéines sur la base de relations empiriques structure/fonction |
| CARA | Affectation de résonance assistée par ordinateur |
| Serveur d'accueil | Un serveur web d'amarrage moléculaire |
| StarBiochem | Un visualiseur de protéines Java, propose une recherche directe dans la banque de données de protéines |
| BÊCHE | L'environnement de développement d'applications de protéomique structurale |
| PocketSuite | Un portail Web pour divers serveurs Web pour l'analyse au niveau du site de liaison. PocketSuite est divisé en : PocketDepth (Prédiction du site de liaison) ; PocketMatch (comparaison des sites de liaison) ; PocketAlign (alignement des sites de liaison) et PocketAnnotate (annotation des sites de liaison). |
| MSL | Une bibliothèque de logiciels de modélisation moléculaire C++ open-source pour la mise en œuvre de méthodes d'analyse structurelle, de prédiction et de conception |
| PSSpred | Prédiction de la structure secondaire des protéines |
| Protée | Outil Web pour suggérer des paires de mutations |
| SDM | Un serveur pour prédire les effets des mutations sur la stabilité des protéines |
Bibliographie
- Bourne PE, Gu J (2009). Structural Bioinformatics (2nd ed.). New York: John Wiley & Sons. (ISBN 978-0-470-18105-8)[40].
- Bourne PE, Weissig H (2003). Structural Bioinformatics. Wiley. (ISBN 0-471-20199-5)[41].
- Leach A (2001). Molecular Modelling: Principles and Applications (2nd ed.). Prentice Hall. (ISBN 978-0-582-38210-7)[42].
- Peitsch MC, Schwede T (2008). Computational Structural Biology: Methods and Applications. World Scientific. (ISBN 978-9812778772)[43].
- Leontis NB, Westhof E (April 2001). "Geometric nomenclature and classification of RNA base pairs". RNA. 7 (4): 499–512. doi:10.1017/S1355838201002515. PMC 1370104. PMID 11345429[44].
- Richardson JS (1981). "The anatomy and taxonomy of protein structure". Advances in Protein Chemistry Volume 34. Advances in Protein Chemistry. Vol. 34. p. 167–339. doi:10.1016/S0065-3233(08)60520-3. (ISBN 978-0-12-034234-1). PMID 7020376[45].
- Ramachandran GN, Sasisekharan V (1968). "Conformation of polypeptides and proteins". Advances in Protein Chemistry Volume 23. Advances in Protein Chemistry. Vol. 23. p. 283–438. doi:10.1016/S0065-3233(08)60402-7. (ISBN 978-0-12-034223-5). PMID 4882249[46].
- Ramachandran GN, Ramakrishnan C, Sasisekharan V (July 1963). "Stereochemistry of polypeptide chain configurations". Journal of Molecular Biology. 7: 95–9. doi:10.1016/S0022-2836(63)80023-6. PMID 13990617[47].
Notes et références
Notes
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Structural bioinformatics » (voir la liste des auteurs).
Références
- Jenny Gu et Philip E. Bourne, Structural Bioinformatics, Hoboken, 2nd, (ISBN 978-1-118-21056-7, OCLC 778339075)
- (en) Jenny Gu et Philip E. Bourne, Structural Bioinformatics, John Wiley & Sons, (ISBN 978-0-470-18105-8, lire en ligne)
- « Comparative visualization of protein secondary structures », BMC Bioinformatics, vol. 18, no Suppl 2, , p. 23 (PMID 28251875, PMCID 5333176, DOI 10.1186/s12859-016-1449-z)
- « Web3DMol: interactive protein structure visualization based on WebGL », Nucleic Acids Research, vol. 45, no W1, , W523–W527 (PMID 28482028, PMCID 5570197, DOI 10.1093/nar/gkx383)
- « Protein-peptide interactions », Current Opinion in Structural Biology, vol. 5, no 1, , p. 103–13 (PMID 7773739, DOI 10.1016/0959-440X(95)80015-S)
- Multifaceted Roles of Crystallography in Modern Drug Discovery, Dordrecht, Springer, coll. « NATO Science for Peace and Security Series A: Chemistry and Biology », , 83–92 p. (ISBN 978-3-642-17906-8, DOI 10.1007/978-3-642-17907-5_4), « Protein-ligand interactions as the basis for drug action. »
- « Proteus | PROTein Engineering Supporter | », proteus.dcc.ufmg.br (consulté le )
- Pedro M. Martins, Vinícius D. Mayrink, Sabrina de A. Silveira et Carlos H. da Silveira, « How to compute protein residue contacts more accurately? », Proceedings of the 33rd Annual ACM Symposium on Applied Computing, ACM, , p. 60–67 (ISBN 978-1-4503-5191-1, DOI 10.1145/3167132.3167136, lire en ligne)
- « Protein cutoff scanning: A comparative analysis of cutoff dependent and cutoff free methods for prospecting contacts in proteins », Proteins, vol. 74, no 3, , p. 727–43 (PMID 18704933, DOI 10.1002/prot.22187, S2CID 1208256, lire en ligne)
- « PDBx/mmCIF General FAQ », mmcif.wwpdb.org (consulté le )
- (en) wwPDB.org, « wwPDB: File Formats and the PDB », www.wwpdb.org (consulté le )
- « PDBx/mmCIF Dictionary Resources », mmcif.wwpdb.org (consulté le )
- « Macromolecular Structures Resource Group », www.ncbi.nlm.nih.gov (consulté le )
- « Nucleic Acid Database (NDB) », ndbserver.rutgers.edu (consulté le )
- « SCOP: Structural Classification of Proteins » [archive du ], (consulté le )
- « Structural alignment of proteins by a novel TOPOFIT method, as a superimposition of common volumes at a topomax point », Protein Science, vol. 13, no 7, , p. 1865–74 (PMID 15215530, PMCID 2279929, DOI 10.1110/ps.04672604)
- « EDS - Uppsala Electron Density Server », eds.bmc.uu.se (consulté le )
- « Home - Prediction Center », www.predictioncenter.org (consulté le )
- « :: Dunbrack Lab », dunbrack.fccc.edu (consulté le )
- « Structural Biology KnowlegebaseSBKB - SBKB », sbkb.org (consulté le )
- « Protein Common Interface Database », dunbrack2.fccc.edu (consulté le )
- « AlphaFold »
- « Structural alignment (genomics) », ScienceDaily (consulté le )
- « Cutoff Scanning Matrix (CSM): structural classification and function prediction by protein inter-residue distance patterns », BMC Genomics, vol. 12 Suppl 4, no S4, , S12 (PMID 22369665, PMCID 3287581, DOI 10.1186/1471-2164-12-S4-S12)
- « A Computational Method to Propose Mutations in Enzymes Based on Structural Signature Variation (SSV) », International Journal of Molecular Sciences, vol. 20, no 2, , p. 333 (PMID 30650542, PMCID 6359350, DOI 10.3390/ijms20020333)
- (en) Kaczanowski et Zielenkiewicz, « Why similar protein sequences encode similar three-dimensional structures? », Theoretical Chemistry Accounts, vol. 125, nos 3–6, , p. 643–650 (ISSN 1432-881X, DOI 10.1007/s00214-009-0656-3, S2CID 95593331, lire en ligne)
- « The relation between the divergence of sequence and structure in proteins », The EMBO Journal, vol. 5, no 4, , p. 823–6 (PMID 3709526, PMCID 1166865, DOI 10.1002/j.1460-2075.1986.tb04288.x)
- « So much more to know », Science, vol. 309, no 5731, , p. 78–102 (PMID 15994524, DOI 10.1126/science.309.5731.78b)
- « Comparative Protein Structure Modeling Using MODELLER », Current Protocols in Bioinformatics, vol. 47, no 1, , p. 5.6.1–32 (PMID 25199792, PMCID 4186674, DOI 10.1002/0471250953.bi0506s47)
- Anupam Dhasmana, Sana Raza, Roshan Jahan, Mohtashim Lohani et Arif, New Look to Phytomedicine, Academic Press, , 523–548 p. (ISBN 978-0-12-814619-4, DOI 10.1016/b978-0-12-814619-4.00020-3, S2CID 69534557), « Chapter 19 - High-Throughput Virtual Screening (HTVS) of Natural Compounds and Exploration of Their Biomolecular Mechanisms: An In Silico Approach »
- The practice of Medicinal Chemistry, Academic Press, , 73–99 p. (ISBN 978-0-12-417205-0, DOI 10.1016/B978-0-12-417205-0.00004-3), « Strategies in the search for new lead compounds or original working hypotheses. »
- « Molecular Dynamics Gives New Insights into the Glucose Tolerance and Inhibition Mechanisms on β-Glucosidases », Molecules, vol. 24, no 18, , p. 3215 (PMID 31487855, PMCID 6766793, DOI 10.3390/molecules24183215)
- (en) « Studies in Molecular Dynamics. I. General Method », The Journal of Chemical Physics, vol. 31, no 2, , p. 459–466 (ISSN 0021-9606, DOI 10.1063/1.1730376, Bibcode 1959JChPh..31..459A)
- Yousif, « Exploring the Molecular Interactions between Neoculin and the Human Sweet Taste Receptors through Computational Approaches », Sains Malaysiana, vol. 49, no 3, , p. 517–525 (DOI 10.17576/jsm-2020-4903-06, lire en ligne)
- MOE
- BALLView
- PyMOL
- VMD
- STRIDE
- Jenny Gu et Philip E. Bourne, Structural bioinformatics, Wiley-Blackwell, (ISBN 978-1-61344-614-0 et 1-61344-614-4, OCLC 799881918, lire en ligne)
- Philip E. Bourne et Helge Weissig, Structural bioinformatics, Wiley-Liss, (ISBN 0-471-20200-2, 978-0-471-20200-4 et 0-471-20199-5, OCLC 50199108, lire en ligne)
- Andrew R. Leach, Molecular modelling : principles and applications, Prentice Hall, (ISBN 0-582-38210-6 et 978-0-582-38210-7, OCLC 45008511, lire en ligne)
- Torsten Schwede et Manuel C. Peitsch, Computational structural biology : methods and applications, World Scientific, (ISBN 981-277-878-0 et 978-981-277-878-9, OCLC 316004177, lire en ligne)
- NEOCLES B. LEONTIS et ERIC WESTHOF, « Geometric nomenclature and classification of RNA base pairs », RNA, vol. 7, no 4, , p. 499–512 (ISSN 1355-8382, DOI 10.1017/s1355838201002515, lire en ligne, consulté le )
- Christian B. Anfinsen, John T. Edsall et Frederic M. Richards, Advances in protein chemistry. Volume 34, Academic Press, (ISBN 978-0-08-058207-8, 0-08-058207-9 et 0-12-034234-0, OCLC 373466726, lire en ligne)
- Christian B. Anfinsen, Advances in protein chemistry. Volume 23, Academic Press, (ISBN 978-0-12-034223-5, 0-12-034223-5 et 978-0-08-058196-5, OCLC 646758501, lire en ligne)
- G. N. Ramachandran, C. Ramakrishnan et V. Sasisekharan, « Stereochemistry of polypeptide chain configurations », Journal of Molecular Biology, vol. 7, , p. 95–99 (ISSN 0022-2836, PMID 13990617, DOI 10.1016/s0022-2836(63)80023-6, lire en ligne, consulté le )
Voir aussi
- MMDB
- Protein Data Bank
- Structural Classification of Proteins database
- STING
- Molecular modelling
- List of software for molecular mechanics modeling
- Molecular Conceptor
- Protein structure prediction
- CASP