Modélisation de protéines par enfilage
La modélisation d'une protéine par enfilage ou modélisation par reconnaissance des repliements est une technique utilisée pour modéliser des protéines dont on souhaite qu'elles présentent les mêmes coudes que des structures de protéines connues, mais qui ne possèdent pas de protéines homologues recensées dans la banque de données sur les protéines (PDB). Elle s'oppose donc à la méthode de prédiction de structure basée sur la modélisation par homologie. La modélisation par enfilage fonctionne en utilisant la connaissance statistique de la relation entre les structures déposées dans la PDB et la séquence de la protéine que l'on souhaite modéliser.
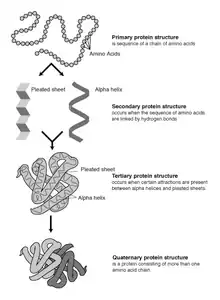
La prédiction est faite en enfilant (c'est-à-dire en plaçant, en alignant) chaque acide aminé dans la séquence cible vers une position dans la structure du modèle et en évaluant dans quelle mesure la cible s'adapte au modèle. Une fois que le modèle le mieux adapté est sélectionné, le modèle structurel de la séquence est construit en fonction de l'alignement sur le modèle choisi.
Une protéine est une structure très fine transversalement mais très longue. Cette structure est le plus souvent repliée sur elle-même pour occuper un espace minimum. L'enveloppe définie par cette structure n'est pas régulière, les interactions des divers champs font que certaines portions de la molécules seront cachées alors que d'autres resteront disponibles pour interagir avec d'autres molécules. Cette forme lui confère des propriétés biologiques additionnelles. Modéliser une protéine, consiste donc en grande partie à essayer de retrouver cette forme 3D. Un élément déterminant de la forme consiste dans ses coudes, c'est-à-dire les endroits où la molécule se replie dans une autre direction.
Cas d'usage
Si on veut inventer une nouvelle protéine, on va sans doute d'abord la définir à partir de ses acides aminés, cependant cela ne suffit pas, son activité biologique est largement définie par sa forme qui est aussi importante que sa composition biochimique. Mais comment connaître sa forme s'il s'agit d'une protéine qui n'existe pas dans la nature ? On peut penser que cette protéine aura une forme similaire à celle qu'aura une protéine dont la composition en acide aminés serait proche.c'est ce que l'on appelle la modélisation de structure par homologie. Cependant il y a des cas de figures où les variations de directions sont rapides et où la méthode de modélisation par homologie donne de mauvais résultats : Les coudes. Il faut donc repérer ces coudes dans des protéines existantes pour pouvoir les prédire dans des protéines nouvellement inventées. Cette méthode peut aussi servir à prédire une forme de la structure uniquement à partir de la prédiction des coudes. On va alors assembler chaque segment issu de la prédiction à ses voisins. Ils sont donc placés en enfilade les uns par rapport aux autres.
Mode opératoire
L'enfilage ou insertion de protéine, également connue sous le nom de reconnaissance de coudes de repliement, est un procédé de modélisation utilisé pour modéliser par analogie, les protéines qui ont les mêmes repliements que des protéines de structures connues, mais qui n'ont pas de protéines homologues de structure connue. Il diffère de la méthode de modélisation de structure par homologie car il est utilisé pour les protéines qui n'ont pas la structure de leurs protéines homologues déposées dans la banque de données protéiques (PDB), alors que la modélisation par homologie est justement utilisée pour les protéines qui ont des protéines homologues déposées dans PDB, et dont on connaît la structure.
La prédiction est faite en « alignant ou en enfilant » chaque acide aminé dans la séquence cible à une position dans une structure servant de gabarit et en évaluant à quel degré le gabarit ainsi « décoré » devient similaire à la séquence cible. Cette opération est répétée sur différents gabarits et finalement on choisit le gabarit qui permet de générer une structure la plus proche possible de celle de la séquence cible. L'enfilage des protéines est basé sur deux observations fondamentales: le nombre de coudes de repliement différents dans la nature est assez faible (environ 1300), et 90 % des nouvelles structures soumises à PDB au cours des trois dernières années présentent des coudes de repliement structurels semblables à ceux déjà contenus dans PDB.
Classification structurale des protéines
La base de données de la classification structurale des protéines (SCOP) fournit une description détaillée et complète des relations structurelles et évolutives de la structure connue. Les protéines sont classées de manière à refléter à la fois la structure et l'évolution. De nombreux niveaux existent dans la hiérarchie, mais les principaux niveaux sont la famille, la superfamille et le pli, comme décrit ci-dessous.
Famille (relation évolutive évidente)
Les protéines regroupées en familles sont clairement liées par l'évolution. En général, cela signifie que les similitudes de résidus par paires entre les protéines sont de 30 % et plus. Cependant, dans certains cas, des fonctions et des structures similaires fournissent des preuves définitives de la descendance commune en l'absence d'identité de séquence élevée. Par exemple, de nombreuses globines forment une famille bien que certains membres aient des identités de séquence de seulement 15 %.
Superfamille (origine évolutionnaire commune probable)
Les protéines qui ont une faible identité de séquence, mais dont les caractéristiques structurelles et fonctionnelles suggèrent qu'une origine évolutive commune est probable, sont placées ensemble dans des superfamilles. Par exemple, l'actine, le domaine ATPase de la protéine de choc thermique et l'hexakinase forment ensemble une superfamille.
Pli (ressemblance structurelle majeure)
Les protéines sont définies comme ayant un pli (coude) commun si elles ont les mêmes structures secondaires majeures dans le même arrangement et avec les mêmes connexions topologiques. Différentes protéines avec le même pli ont souvent des éléments périphériques de structure secondaire et des régions de coude qui diffèrent en taille et en conformation. Dans certains cas, ces régions périphériques différentes peuvent couvrir la moitié de la structure. Les protéines placées dans la même catégorie de coudes de repliement peuvent ne pas avoir une origine évolutive commune: les similitudes structurelles pourraient provenir uniquement de la physique et de la chimie des protéines favorisant certains arrangements d'emballage et la topologie de chaîne.
Méthode
Un paradigme général d'enfilage de protéine se compose des quatre étapes suivantes :
- La construction d'une base de données de modèles de structure : Il faut sélectionner des structures de protéines à partir des bases de données de structures de protéines. On va utiliser ces structures de protéines comme modèles structuraux. Cela implique généralement de sélectionner des structures de protéines dans des bases de données telles que PDB, FSSP, SCOP ou CATH, après avoir éliminé les protéines qui présentent des similitudes de séquence élevées car cela introduirait un biais dans la modélisation.
- La conception de la fonction de notation : Il faut concevoir une fonction de notation qui va mesurer l'adéquation entre les séquences cibles et les modèles qui sont générés en « décorant » les gabarits, en fonction de la connaissance des relations connues entre les structures et les séquences. Une bonne fonction de notation doit contenir le potentiel de mutation, le potentiel de conditionnement physique de l'environnement, le potentiel d’appairage, les compatibilités de structure secondaire et les pénalités d'écart. La qualité de la fonction d'énergie est étroitement liée à la précision de prédiction, en particulier la précision d'alignement.
- Alignement de la séquence cible : Il faut aligner la séquence cible avec chacun des modèles de structure en optimisant le résultat fournit par la fonction de notation. Cette étape est l'une des tâches principales de tous les programmes de prédiction de structure basés sur l’enfilage de protéine qui tiennent compte du potentiel de contact par appairage. Sinon, un algorithme de programmation dynamique suffit.
- Prédiction d'enfilage de protéine : Il faut sélectionner l'alignement des enfilages de protéine qui est statistiquement le plus probable. Ensuite, il faut construire un modèle de structure pour la cible en plaçant les atomes d’armature de la séquence cible à leurs positions d’armature alignées pour le modèle de structure sélectionné.
Comparaison avec la modélisation par homologie
La modélisation par homologie et enfilage des protéines sont des méthodes à base de modèles et il n'y a pas de limite rigoureuse entre elles en termes de techniques de prédiction. Mais les structures protéiques de leurs cibles sont différentes. La modélisation par homologie ne sera possible que pour les cibles qui ont des protéines homologues de structure connue, tandis que l’enfilage de protéines n’est possible que pour les cibles avec seulement l'homologie de coudes de repliement trouvé. En d'autres termes, la modélisation par homologie est destinée à des cibles «plus faciles» et l’enfilage des protéines est destinée à des cibles « plus difficiles ».
La modélisation par homologie traite le modèle dans un alignement en tant que séquence, et seule l'homologie de séquence est utilisée pour la prédiction. L'enfilage de protéines traite le gabarit dans un alignement en tant que structure, et les informations de séquence et de structure extraites de l'alignement sont utilisées pour la prédiction. Lorsqu'il n'y a pas d'homologie significative trouvée, l'enfilage de protéine peut faire une prédiction basée sur l'information de structure. Cela explique également pourquoi l'enfilage de protéine peut être plus efficace que la modélisation homologie dans de nombreux cas.
En pratique, lorsque l'identité de séquence dans un alignement de séquences de séquences est faible (c'est-à-dire <25 %), la modélisation par homologie peut ne pas produire une prédiction significative. Dans ce cas, s'il existe une homologie lointaine pour la cible, la modélisation par enfilage de la protéine peut générer une bonne prédiction.
Logiciels d'enfilage de protéines
- HHpred est un serveur d'enfilage très répandu qui exécute HHsearch, un logiciel largement utilisé pour la détection d'homologie à distance, basé sur une comparaison par paire de modèles de Markov cachés.
- Raptor (logiciel) est un logiciel d'enfilage de protéines basé sur la programmation par nombres entiers. Il a été remplacé par un nouveau programme d'enfilage de protéines, RaptorX. Ce logiciel de modélisation et d'analyse de protéines utilise des modèles graphiques probabilistes et une inférence statistique pour les threads de protéines à matrice unique et multi-matrice[1] - [2] - [3] - [4]. RaptorX surpasse de manière significative RAPTOR et est particulièrement doué pour l’alignement de protéines avec un profil de séquence clairsemé. Le serveur RaptorX est gratuit pour le public.Phyre est un serveur d'enfilage populaire associant HHsearch à une modélisation ab initio et à modèles multiples.
- Muster est un algorithme d'enfilage standard basé sur la programmation dynamique et l'alignement de profil de séquence. Il combine également plusieurs ressources structurelles pour faciliter l'alignement du profil de séquence[5].
- Sparks X est une correspondance probabiliste séquence-structure structurelle entre les propriétés structurelles prédictives unidimensionnelles de la requête et les propriétés natives correspondantes des modèles[6].
- BioShell est un algorithme d'enfilage utilisant un algorithme de programmation dynamique optimisé de profil à profil combiné à une structure secondaire prédite[7].
Notes et références
- (en) Jian Peng et Jinbo Xu, « Raptorx: Exploiting structure information for protein alignment by statistical inference », Proteins: Structure, Function, and Bioinformatics, vol. 79, no S10, , p. 161–171 (DOI 10.1002/prot.23175, lire en ligne, consulté le )
- (en) J. Peng et J. Xu, « Low-homology protein threading », Bioinformatics, vol. 26, no 12, , i294–i300 (ISSN 1367-4803 et 1460-2059, PMID 20529920, PMCID PMC2881377, DOI 10.1093/bioinformatics/btq192, lire en ligne, consulté le )
- (en) Jian Peng et Jinbo Xu, « A multiple-template approach to protein threading », Proteins: Structure, Function, and Bioinformatics, vol. 79, no 6, , p. 1930–1939 (DOI 10.1002/prot.23016, lire en ligne, consulté le )
- (en) Jianzhu Ma, Jian Peng, Sheng Wang et Jinbo Xu, « A conditional neural fields model for protein threading », Bioinformatics, vol. 28, no 12, , i59–i66 (ISSN 1460-2059 et 1367-4803, PMID 22689779, PMCID PMC3371845, DOI 10.1093/bioinformatics/bts213, lire en ligne, consulté le )
- (en) Sitao Wu et Yang Zhang, « Muster: Improving protein sequence profile-profile alignments by using multiple sources of structure information », Proteins: Structure, Function, and Bioinformatics, vol. 72, no 2, , p. 547–556 (DOI 10.1002/prot.21945, lire en ligne, consulté le )
- (en) Y. Yang, E. Faraggi, H. Zhao et Y. Zhou, « Improving protein fold recognition and template-based modeling by employing probabilistic-based matching between predicted one-dimensional structural properties of query and corresponding native properties of templates », Bioinformatics, vol. 27, no 15, , p. 2076–2082 (ISSN 1367-4803 et 1460-2059, PMID 21666270, PMCID PMC3137224, DOI 10.1093/bioinformatics/btr350, lire en ligne, consulté le )
- (en) D. Gront, M. Blaszczyk, P. Wojciechowski et A. Kolinski, « BioShell Threader: protein homology detection based on sequence profiles and secondary structure profiles », Nucleic Acids Research, vol. 40, no W1, , W257–W262 (ISSN 0305-1048 et 1362-4962, PMID 22693216, PMCID PMC3394251, DOI 10.1093/nar/gks555, lire en ligne, consulté le )
Bibliographie
- (en) AV Finkelstein et BA Reva, « A search for the most stable folds of protein chains », Nature, vol. 351, no 6326, , p. 497–9 (PMID 2046752, DOI 10.1038/351497a0)
- (en) Lathrop RH, « The protein insertion problem with sequence amino acid interaction preferences is NP-complete », Protein Eng, vol. 7, no 9, , p. 1059–1068 (PMID 7831276, DOI 10.1093/protein/7.9.1059)
- (en) D. T. Jones et C. Hadley, Bioinformatics: Sequence, structure and databanks, Heidelberg, Springer-Verlag, , 1–13 p., « insertion methods for protein structure prediction »
- (en) J. Xu, M. Li, D. Kim et Y. Xu, « Raptor: Optimal Protein insertion by Linear Programming, the inaugural issue », J Bioinform Comput Biol, vol. 1, no 1, , p. 95–117 (PMID 15290783, DOI 10.1142/S0219720003000186)
- (en) J. Xu, M. Li, G. Lin, D. Kim et Y. Xu, « Protein insertion by linear programming », Pac Symp Biocomput, , p. 264–275 (PMID 12603034)