Tyrosine hydroxylase
La tyrosine hydroxylase est une oxydoréductase qui catalyse la réaction :
- L-tyrosine + tétrahydrobioptérine + O2 L-DOPA + 4a-hydroxytétrahydrobioptérine.

| N° EC | EC |
|---|---|
| N° CAS | |
| Cofacteur(s) | Fe2+ |
| IUBMB | Entrée IUBMB |
|---|---|
| IntEnz | Vue IntEnz |
| BRENDA | Entrée BRENDA |
| KEGG | Entrée KEGG |
| MetaCyc | Voie métabolique |
| PRIAM | Profil |
| PDB | RCSB PDB PDBe PDBj PDBsum |
| GO | AmiGO / EGO |
Cette enzyme convertit la tyrosine en L-DOPA, ou dihydroxyphénylalanine.
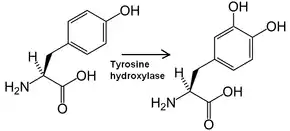
Elle utilise pour cela de l'oxygène O2 ainsi qu'un cation de fer Fe2+ ainsi que de la tétrahydrobioptérine comme cofacteurs. La L-DOPA est un précurseur de la dopamine, qui est elle-même un précurseur important de la noradrénaline et de l'adrénaline. Elle catalyse également l'étape limitante de la biosynthèse des catécholamines. Chez l'homme, cette enzyme est encodée par le gène TH et est présente dans le système nerveux central, les neurones sympathiques périphériques la médullosurrénale[1]. La tyrosine hydroxylase fait partie, avec la phénylalanine hydroxylase et la tryptophane hydroxylase, de la famille des hydroxylases d'acides aminés aromatiques (AAAH) à bioptérine.
Structure et caractéristiques de la tyrosine hydroxylase
La tyrosine hydroxylase (TH), aussi appelée tyrosine-3-monooxygénase, appartient à la famille des hydroxylases des acides aminés aromatiques. Elles sont dépendantes de la tétrahydrobioptérine et du fer mais leur structure n’est pas hème. En effet, la TH nécessite un cofacteur naturel donneur d’hydrogène, la tétrahydrobioptérine (H4BPt), synthétisée à partir de la guanosine 5’ triphosphate dans les cellules possédant l’enzyme tyrosine hydroxylase. La tyrosine hydroxylase possède dans sa structure un site actif, du fer (II) qui est relié à deux histidines ainsi qu’à plusieurs ligands qui possèdent de l’oxygène, comme de l’eau ; et ce selon une structure hexagonale[2]. La tyrosine hydroxylase a une masse atomique de 240 kilodaltons et est formée de quatre sous-unités identiques d’environ 60 kDa. D’après la séquence de nucléotides, il a été établi que le gène de la tyrosine hydroxylase compte environ 8,5 kilobases et est formé de 14 exons séparés par 12 introns. Cette enzyme est présente sous quatre formes chez l’humain tandis qu’elle existe uniquement sous une forme chez les animaux. L’ARN messager de ces quatre isoenzymes sont majoritairement identiques sauf près de l’extrémité N-terminale. Ces différences sont dues à des insertions et des délétions spécifiques. Cette enzyme est cytoplasmique et on la retrouve à des endroits précis dans le cerveau, soit dans la substance noire, dans le locus coeruleus et dans la médullaire surrénale[3].
La tyrosine hydroxylase est formée de deux domaines, soit un catalyseur et un régulateur. Le domaine catalyseur est situé près de l’extrémité C-terminale et est caractérisé par la présence d’acides aminés conservés à travers les espèces. En effet, ce domaine contient sept cystéines chez l’humain et six de ces sept cystéines sont présentes chez les animaux. La cystéine permet le maintien de la conformation de la tyrosine hydroxylase car elle forme des ponts disulfures et interagit avec le fer. Le domaine régulateur est plutôt situé à l’extrémité N-terminale et possède un effet inhibiteur sur l’activité de l’enzyme. Il a aussi été démontré que la phosphorylation et la déphosphorylation de la tyrosine hydroxylase régulent l’activité de cette enzyme. Cette régulation affecte plus particulièrement l’activité dopaminergique nigro-striée. La phosphorylation est effectuée par différentes protéines kinases et on dénombre pour le moment cinq sites de phosphorylation situés à l’extrémité N-terminale : sérine-8, sérine-19, sérine-31, sérine-40 et sérine-153. La phosphorylation de chacun de ces sites est effectuée par différentes enzymes et selon différents mécanismes. Puisque la phosphorylation joue un rôle important dans la régulation de l’activité de la tyrosine hydroxylase, il pourrait être possible d’augmenter la synthèse de dopamine en augmentant cette phosphorylation. Ceci pourrait s’effectuer en activant une ou plusieurs kinases tout en inhibant une ou plusieurs phosphatases, des enzymes qui déphosphorylent la tyrosine hydroxylase. Ceci a d’ailleurs été observé à l’aide de l’acide okadaic, un inhibiteur de phosphatases, ce qui a permis d’observer une augmentation de l’activité de la TH dans des cultures cellulaires. Cet acide est par contre inutilisable chez l’humain car il est cancérigène[3].
Rôle de la tyrosine hydroxylase dans la synthèse des catécholamines
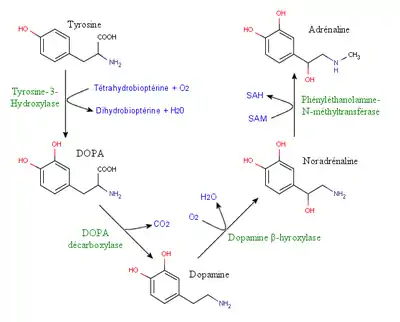
Les catécholamines sont des composés organiques qui sont synthétisés à partir d’un acide aminé aromatique et polaire, la L-tyrosine. La dopamine, l’épinéphrine et la norépinéphrine sont les trois composés les plus connus de ce groupe de molécules et peuvent agir comme un neurotransmetteur ou comme une hormone. Ces trois amines sont synthétisées selon une chaîne métabolique commune et sont donc considérées comme des molécules liées l’une à l’autre. La L-tyrosine est la molécule qui joue le rôle de précurseur de la séquence métabolique. Cet acide aminé est retrouvé de façon abondante dans la nourriture et peut être également synthétisé à partir de la phénylalanine[4]. Cette molécule se retrouve donc dans la circulation sanguine et traverse ensuite la barrière hémato-encéphalique pour y être captée par des neurones catécholaminergiques par l’entremise du système de transport des acides aminés neutres. Ce type de transport est de type symport car il est couplé au transport des ions Na+[5].
La tyrosine hydroxylase joue un rôle essentiel dans le métabolisme de la L-tyrosine et donc dans la synthèse des catécholamines. En effet, cette enzyme cytoplasmique, à la suite de son activation par la phosphorylation produite par une protéine kinase A, catalyse la transformation de la L-tyrosine en L-DOPA (DihydrOxyPhénylAlanine). Cette réaction est irréversible et nécessite la présence de fer ferreux, d’oxygène respiratoire ainsi que de tétrahydrobioptérine (H4BPt). Les atomes d'hydrogène réduisent alors un atome d’oxygène tandis qu’un second atome d’oxygène vient se lier sur le troisième carbone du noyau aromatique de la phénylalanine[6]. La L-DOPA produite est ensuite convertie en dopamine par l’enzyme dopa-décarboxylase, qui est ensuite transformée en norépinéphrine (noradrénaline) par l’enzyme dopamine-bêta-hydroxylase et qui est finalement elle-même convertie en épinéphrine (adrénaline) par l’enzyme phényléthanolamine-N-méthyltransférase. Ces transformations successives confirment donc que la synthèse des catécholamines est une voie commune et dépendent alors l’une de l’autre[4]. Un fait intéressant dans la synthèse de ces molécules est que l’enzyme tyrosine hydroxylase est l’étape limitante de ce processus car elle est présente en quantité limitée. C’est donc la quantité et l’activité de cette enzyme qui déterminent la quantité de catécholamines produites et ce, sans lien avec la quantité de tyrosine présente[4].L’activité de la tyrosine hydroxylase est également régulée par de nombreux facteurs. Par exemple, les produits terminaux de synthèse comme l’épinéphrine et la norépinéphrine peuvent agir comme « feed back » négatif[5]. Ces divers facteurs limitants de synthèse peuvent être contournés en ingérant de façon orale de la L-DOPA. On applique d’ailleurs cette technique comme médicament utilisé comme traitement chez les patients souffrant de la maladie de Parkinson[4].
La tyrosine hydroxylase et la maladie de Parkinson
La maladie de Parkinson est une affection neurodégénérative caractérisée par une atteinte de la coordination et de la motricité volontaire. Elle apparaît habituellement après la cinquantaine. On ne connaît pas encore la cause de cette maladie et on ne peut donc pas la guérir. Les symptômes les plus courants sont les tremblements, des bradykinésies ainsi qu’une rigidité musculaire. Au Canada, on estime que 100 000 personnes sont atteintes de Parkinson. De ce nombre, on compte environ 25 000 Québécois atteints[7]. De plus, ce nombre tend à augmenter avec le vieillissement de la population[8]. Les statistiques démontrent d’ailleurs que 1 % de la population âgée de 65 ans et plus ainsi que 2 % des gens de 70 ans et plus seront atteints du Parkinson[7].
La maladie de Parkinson est caractérisée par une diminution de neurones dopaminergiques dans la substance noire du mésencéphale ainsi que par une baisse du contrôle de la voie nigro-striée, soit la voie du mésencéphale vers le striatum. La dopamine est sécrétée par les neurones de la substance noire et ceux-ci sont reliés à d’autres neurones présents dans le striatum. Les neurones du striatum transmettent ensuite les messages de commandes de mouvement au cortex, qui les transmet finalement aux fibres musculaires. La diminution de neurones dopaminergiques dans la substance noire se traduit par un déficit en dopamine. Cette insuffisance provoque donc une baisse des signaux transmis au striatum entraînant alors une baisse de messages envoyés via la voie nigro-striée. On observe finalement une diminution de signaux envoyés vers le cortex à partir du striatum et donc une perturbation de l’appareil moteur[9]. Au moment du diagnostic, on compte qu’environ 80 % des neurones produisant de la dopamine ont cessé de fonctionner[10].
Dans 95 % des cas, on ne connaît pas les causes de la maladie de Parkinson. En effet, seulement 5 % de la maladie est due à l’hérédité. On croit par contre que cette maladie serait peut-être due à un stress oxydant ou à une neuroinflammation. Le stress oxydant est provoqué par des radicaux libres endogènes synthétisés lors de la respiration cellulaire ou par des radicaux libres exogènes provenant de pesticides et d’insecticides. Ces radicaux libres modifient le potentiel membranaire de la mitochondrie et bloque alors la chaîne de transport d’électrons. Ce phénomène provoque l’accumulation de radicaux superoxydes qui entraînent l’apoptose de la cellule, soit des neurones dopaminergiques.
De plus, on a observé une diminution de l’activité de tyrosine hydroxylase dans la voie nigro-striée des gens atteints du Parkinson. Cette baisse de TH serait due à la diminution des neurones dopaminergiques dans la substance noire et dans le striatum[11]. Il a également été suggéré que TH contribuerait à la production de radicaux libres comme du H2O2, provoquant ainsi du stress oxydant[2].
Identité de tyrosine hydroxylase dans des banques de données
- EntrezProtein:AAI04968
- OMIM:191290
- Structure:1TOH
Notes et références
- (en) T. Nagatsu, « Tyrosine hydroxylase: human isoforms, structure and regulation in physiology and pathology », Essays in Biochemistry, vol. 30, , p. 15-35 (PMID 8822146)
- Haavik, J., Toska, K., Tyrosine Hydroxylase and Parkinson’s Disease. Molecular Neurobiology, 1998. 16(3) :p. 285-309.
- Goldstein, M., Lieberman, A., The role of the regulatory enzymes of catecholamine synthesis in Parkinson’s disease. NEUROLOGY, 1992. 42(4) :p. 8-10.
- Kolb, B., Whishaw, I. Cerveau & comportement, De Boeck & Larcier, 2002, Paris, 646 pages.
- http://neurobranches.chez-alice.fr/neurophy/catecholamines.html, Les catécholamines
- http://www.chups.jussieu.fr/polys/biochimie/CNbioch/POLY.Chp.9.3.html, Tyrosine hydroxylase
- http://www.parkinsonquebec.ca/fr/mal_quitouche.htm, Qui en est atteint?
- http://www.hc-sc.gc.ca/ahc-asc/minist/health-sante/messages/2007_04_05_f.html, Mois de sensibilisation à la maladie de Parkinson – 2007
- http://www.phac-aspc.gc.ca/publicat/cdic-mcc/20-2/b_f.html, Série de monographies sur les maladies liées au vieillissement : XII. Maladie de Parkinson - Percées récentes et nouvelles orientations
- http://www.parkinsonquebec.ca/fr/mal_questce.htm, Le Parkinson, qu’est-ce que c’est?
- Mogi,. M., Harada, M., Kiuchi, K., Kojima, K., Kondo, T., Narabayashi, H., Rausch, D., Riederer, P., Jellinger, K., Nagatsu, T., Homospecific activity (activity per ensyme protein) of tyrosine hydroxylase increases in Parkinsonian brain. J Neural Transm, 1988. 72 :p. 77-81.