Rétrocoordination π
En chimie de coordination, une rétrocoordination π (π backbonding ou π back-donation en anglais) est un type de liaison chimique caractérisé par la cession d'électrons d'une orbitale atomique d'un métal vers une orbitale antiliante π* sur un ligand π-accepteur[1] - [2]. Elle est particulièrement courante dans les complexes organométalliques de métaux de transition avec des ligands polyatomiques tels que le monoxyde de carbone CO ou l'éthylène CH2=CH2. Les électrons du métal interviennent pour se lier au ligand en libérant le métal d'un excès de charge électrique négative. Le tétracarbonyle de nickel Ni(CO)4 et le sel de Zeise K[PtCl3(C2H4)]·H2O sont des complexes présentant des rétrocoordinations π.
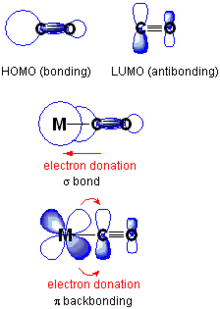
Au centre : exemple d'orbitale σ liante dans laquelle le CO cède des électrons de son orbitale HO au métal.
En bas : exemple dans lequel le métal cède des électrons via une orbitale d à l'orbitale BV du CO.
L'IUPAC donne la définition suivante de la rétrocoordination[3] :
« Une description de la liaison de ligands π-conjugués à un métal de transition faisant intervenir une synergie entre d'une part la cession d'électrons de l'orbitale π occupée ou de l'orbitale de doublet non liant du ligand vers une orbitale inoccupée du métal (liaison donneur-accepteur), et d'autre part la cession (rétrocoordination) d'électrons d'une orbitale nd du métal (de symétrie π par rapport à l'axe métal-ligand) vers l'orbitale antiliante π* vacante du ligand. »
Une rétrocoordination se produit lorsque l'atome métallique présente un excès de charge issu des liaisons σ avec les ligands qu'il compense en cédant des électrons des orbitales d à un ligand π-accepteur. De tels ligands π-accepteurs sont par exemple l'ion cyanure CN−, le monoxyde de carbone CO, l'ion fulminate CNO−, les isonitriles R-N≡C, l'ion acétylure C≡C2− ou encore le cation nitrosonium N≡O+. La rétrocoordination s'observe notamment avec les métaux de transition à un état d'oxydation inférieur ou égal à zéro.
Carbonyles de métaux, nitrosyles et isonitriles
Dans ce type de complexes, les électrons sont partiellement transférés d'une orbitale d du métal vers les orbitales antiliantes du monoxyde de carbone CO et de ces analogues. Ce transfert d'électrons a pour effet de renforcer la liaison carbone–métal et d'affaiblir la liaison carbone–oxygène. Le renforcement de la liaison M–CO se traduit par l'augmentation de la fréquence de vibration de la liaison métal–carbone, souvent hors du champ de la spectroscopie infrarouge habituelle, ainsi que par le raccourcissement de cette liaison. L'affaiblissement de la liaison carbone–oxygène se traduit par la diminution du nombre d'onde, c'est-à-dire par l'augmentation de la longueur d'onde, des bandes νCO par rapport à celles du CO libre : il passe ainsi de 2 143 cm−1 pour le CO libre à 2 060 cm-1 pour le tétracarbonyle de nickel Ni(CO)4, à 1 981 cm-1 pour l'hexacarbonyle de chrome Cr(CO)6 et à 1 790 cm-1 dans l'anion [Fe(CO)4]2−[4]. Pour cette raison, la spectroscopie infrarouge est une technique importante de diagnostic technique en chimie des carbonyles de métaux.
Une rétrocoordination π forte peut s'établir avec de nombreux autres ligands que le monoxyde de carbone. Le monoxyde d'azote NO est un π-accepteur encore plus fort que ce dernier, le nombre d'onde νNO étant un outil de diagnostic utilisé en chimie des nitrosyles de métaux.
Contrairement au monoxyde de carbone, le doublet non liant σ-donneur de l'atome de carbone des isonitriles R–N≡C est naturellement antiliant ; la liaison C≡N est renforcée par la complexation, et le nombre d'onde νCN est augmenté. Cependant, la rétrocoorination π diminue νCN. Selon le rapport des forces entre la liaison σ et la rétrocoordination π, νCN peut être augmenté, comme lors de la complexation de métaux faiblement π-donneurs comme Pt(II), ou réduit, comme lors de la complexation avec des π-donneurs forts comme Ni(0)[5]. L'angle MC≡N–C est un autre paramètre entrant en ligne de compte avec les isonitriles, s'écartant de 180° dans les systèmes très riches en électrons.
Les autres ligands donnent une faible rétrocoordination π qui rend les ligands CO labiles, ce qu'on appelle l'effet cis.
Complexes métal–alcène et métal–alcyne
Comme dans le cas des carbonyles de métaux, des électrons sont partiellement transférés d'une orbitale d du métal vers des orbitales antiliantes des alcènes ou des alcynes. Ce transfert d'électrons a pour effet de renforcer la liaison métal–ligand et d'affaiblir les liaisons carbone–carbone au sein des ligands eux-mêmes. Le renforcement des liaisons M–C2R4 et M–C2R2 se traduit par la courbure de l'angle C–C–R, en raison de l'accroissement du caractère respectivement sp3 et sp2. Une forte rétrocoordination π conduit un complexe métal–alcène à adopter des propriétés d'un métallocyclopropane. Les substituants électronégatifs donnent une rétrocoordination π plus forte, de sorte que le tétrafluoroéthylène C2F4, le tétracyanoéthylène C2(CN)4 et l'hexafluoro-2-butyne CF3–C≡C–CF3 sont les ligands présentant une forte rétrocoordination π.
Complexes métal–phosphine
Les phosphines acceptent des électrons d'orbitales p ou d d'un métal dans des orbitales antiliantes σ* de liaisons carbone–phosphore ayant une symétrie π[6]. Lorsque des phosphines se lient à des atomes métalliques riches en électrons, les liaisons carbone–phosphore devraient s'allonger dans la mesure où les orbitales σ* correspondantes se remplissent d'électrons. Cet effet est cependant souvent masqué par le fait que le doublet non liant du phosphore est cédé au métal, ce qui réduit la répulsion provoquée par ce doublet et donc tend à raccourcir cette liaison. Ces deux effets peuvent être résolus en comparant la structure de complexes métal–phosphine ne différant que par un électron[7]. L'oxydation de complexes M–PR3 conduit à des liaisons M–P plus longues et des liaisons P–C plus courtes, ce qui est cohérent avec la rétrocoordination π[8]. Les premiers travaux avaient conduit à penser que les ligands phosphine utilisent leurs orbitales 3d pour établir une rétrocoordination π métal–phosphine, mais il est désormais accepté que les orbitales d du phosphore ne sont pas impliquées dans cette liaison car leur énergie est trop élevée[9] - [10].
Notes et références
- (en) Gary L. Miessler et Donald Arthur Tarr, Inorganic Chemistry, 1999, p. 338. (ISBN 978-0-1384-1891-5).
- (en) Frank Albert Cotton, Geoffrey Wilkinson et Carlos A. Murillo, Advanced Inorganic Chemistry, 1999. (ISBN 978-0-4711-9957-1).
- (en) « back donation », IUPAC, Compendium of Chemical Terminology [« Gold Book »], Oxford, Blackwell Scientific Publications, 1997, version corrigée en ligne : (2019-), 2e éd. (ISBN 0-9678550-9-8) :
« A description of the bonding of π-conjugated ligands to a transition metal which involves a synergic process with donation of electrons from the filled π-orbital or lone electron pair orbital of the ligand into an empty orbital of the metal (donor–acceptor bond), together with release (back donation) of electrons from an nd orbital of the metal (which is of π-symmetry with respect to the metal–ligand axis) into the empty π*-antibonding orbital of the ligand. »
- (en) C. E. Housecroft et A. G. Sharpe, Inorganic Chemistry, Pearson Prentice-Hall, 2e édition, 2005, p. 702. (ISBN 0-130-39913-2).
- (en) Robert H Crabtree, The Organometallic Chemistry of the Transition Metals, Wiley, 6e édition, 2014, p. 105–106. (ISBN 978-1-11813807-6).
- (en) A. Guy Orpen, Neil G. Connelly, « Structural systematics: the role of P-A .sigma.* orbitals in metal-phosphorus .pi.-bonding in redox-related pairs of M-PA3 complexes (A = R, Ar, OR; R = alkyl) », Organometallics, vol. 9, no 4, , p. 1206-1210 (DOI 10.1021/om00118a048, lire en ligne)
- (en) Robert H. Crabtree, The Organometallic Chemistry of the Transition Metals, Wiley, 5e édition, 2009, pp. 99–100. (ISBN 978-0-470-25762-3).
- (en) Barry J. Dunne, Richard B. Morris et A. Guy Orpen, « Structural systematics. Part 3. Geometry deformations in triphenylphosphine fragments: a test of bonding theories in phosphine complexes », Journal of the Chemical Society, Dalton Transactions, vol. 0, no S, , p. 653-661 (DOI 10.1039/DT9910000653, lire en ligne)
- (en) Declan G. Gilheany, « No d Orbitals but Walsh Diagrams and Maybe Banana Bonds: Chemical Bonding in Phosphines, Phosphine Oxides, and Phosphonium Ylides », Chemical Reviews, vol. 94, no 5, , p. 1339-1374 (PMID 27704785, DOI 10.1021/cr00029a008, lire en ligne)
- (en) Natalie Fey, A. Guy Orpen, « Building ligand knowledge bases for organometallic chemistry: Computational description of phosphorus(III)-donor ligands and the metal–phosphorus bond », Coordination Chemistry Reviews, vol. 253, nos 5-6, , p. 704-722 (DOI 10.1016/j.ccr.2008.04.017, lire en ligne)