Procédé Wacker
Le procédé Wacker ou le procédé Hoechst-Wacker (nommé d'après les entreprises chimiques du même nom) est un procédé industriel de type chimique. Il se réfère à l'oxydation de l'éthylène en acétaldéhyde en présence de chlorure de palladium(II) comme catalyseur[1]. Cette réaction chimique a été l'une des premières catalyses homogènes avec un organopalladium appliquée à une échelle industrielle[2].
Histoire
Le développement du procédé Wacker commence en 1956, à la société Wacker Chemie[3]. À l'époque, de nombreux composés industriels sont produits à partir de l'acétylène, dérivé du carbure de calcium, un procédé coûteux et peu respectueux de l'environnement. Quand Esso construit une nouvelle raffinerie de pétrole à Cologne, près de leur site, Wacker se rend compte que l'éthylène serait moins cher que les matières premières. Wacker décide alors d'enquêter sur ses utilisations potentielles. Durant leur recherche pour trouver de l'oxyde d'éthylène, une réaction éthylène et oxygène en présence de charbon palladié forme de l'acétaldéhyde ou éthanal. La découverte est faite grâce à l'odeur caractéristique de pomme de ce composé. Après des recherches sur la transformation de éthylène en éthanal conversion, Wacker obtient un brevet en 1957. Celui-ci décrit une réaction en phase gazeuse en présence d'un catalyseur hétérogène[4]. En même temps, Hoechst AG rejoint la course avec un dépôt de brevet et entraine Wacker dans un partenariat appelé Aldehyd GmbH. Le procédé hétérogène finalement échoue à cause du catalyseur qui ne s'active pas. Le procédé est remplacé par un autre, un système homogène à base d'eau. En 1958, une usine pilote ouvre. L'adoption du titane (nouvellement disponible pour l'utilisation industrielle) comme matériau de construction pour les réacteurs et les pompes résolvent les problèmes avec l'agressivité du catalyseur. Les usines de production entrent en service en 1960.
Mécanisme réactionnel
Le mécanisme réactionnel du procédé industriel Wacker (oxydation de l'oléfine par le chlorure de palladium(II)) a reçu beaucoup d'attention depuis plusieurs décennies. Certains aspects de ce mécanisme font encore l'objet de débat. Une formulation moderne est décrite ci-dessous :
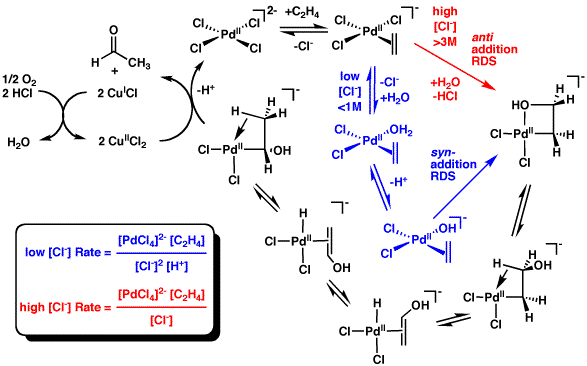
La première réaction stœchiométrique a été rapportée par Phillips[5] - [6]. La réaction peut également être décrite comme :
- [PdCl4]2 − + C2H4 + H2O → CH3CHO + Pd + 2 HCl + 2 Cl−
Cette conversion est suivie par des réactions pour régénérer le catalyseur Pd :
- Pd + 2 CuCl2 + 2 Cl− → [PdCl4]2− + 2 CuCl
- 2 CuCl + ½ O2 + 2 HCl → 2 CuCl2 + H2O
Tous les catalyseurs sont régénérés et seuls l'alcène et l'oxygène sont consommés. Sans chlorure de cuivre(II) (CuCl2) comme agent oxydant, le métal Pd(0) (résultant de l'élimination réductrice de Pd(II) dans l'étape finale) précipiterait, et la réaction s'arrêterait après un cycle. Cette réaction stœchiométrique a été découverte en 1894. De l'air, de l'oxygène pur, ou un certain nombre d'autre oxydants peuvent alors oxyder le chlorure de cuivre(I) (CuCl) en CuCl2, permettant au cycle de continuer.
La réaction Wacker a d'abord été rapportée par Smidt et al.[7] - [8] - [9].
Étude des mécanismes historiques
Des études du mécanisme des années 1960 ont élucidé plusieurs points clés[10] - [11] :
- il n'y a pas d'échange H/D observé dans cette réaction. Des expériences à l'aide de C2D4 en solution génèrent CD3CDO, et les essais avec C2H4 dans D2O génèrent CH3CHO. Ainsi, une tautomérisation céto-énolique n'est pas possible ;
- l'effet isotopique cinétique avec des réactifs entièrement deutérés est négligeable (kH/kD=1,07). On en déduit que le transfert de l'hydrure n'est pas une étape cinétiquement déterminante ;
- un effet isotopique concurrentiel important avec C2H2D2 (kH/kD= ~1,9) suggère que l'étape cinétiquement déterminante doit être avant la formation du produit oxydé ;
- de fortes concentrations de chlorure et de chlorure de cuivre(II) favorisent la formation d'un nouveau produit, la chlorohydrine, qui est une halogénohydrine.
Sur la base de ces observations, il est généralement admis que l'étape cinétiquement déterminante se produit avant une série de réarrangements des hydrures. De nombreuses expériences et de recherches théoriques ont cherché à identifier cette étape, mais sans résultat pour l'instant.
La plupart des études mécanistiques sur le procédé Wacker ont cherché à savoir si l'attaque nucléophile se fait par l'extérieur (addition anti) ou par l'intérieur (addition syn). Grâce à des expériences cinétiques, Henry déduit que le mécanisme de l'attaque nucléophile serait une addition syn. Ensuite, des études stéréochimiques effectuées par Stille et ses collaborateurs[12] - [13] - [14] ont abouti à des produits chimiques qui ont indiqué que le procédé Wacker se produisait par addition anti. Cependant, comme ces expériences ont été menées dans des conditions sensiblement différentes que les conditions industrielles de Wacker, leurs conclusions sont contestées. Les études stéréochimiques modernes recrééant les conditions industrielles (à l'exception des concentrations élevées de chlorure et de chlorure de cuivre) ont également abouti à des produits qui démontraient une addition anti[15]. À la suite de la publication des résultats de deux études indépendantes avec ces résultats, la communauté scientifique a accepté le mécanisme de l'addition anti comme étant la norme du procédé Wacker.
Des études cinétiques en conditions industrielles ont été réalisées sur des alcools allyliques isotopement substitués(avec de faibles concentrations de chlorure) pour sonder les mécanismes de réaction[16] - [17]. Ces résultats ont montré que l'attaque nucléophile était un mécanisme lent, tandis que les mécanismes proposés pour expliquer les études stéréochimiques menées auparavant partaient du principe que l'attaque nucléophile était un mécanisme rapide.
Cependant, les recherches menées ultérieurement ont indiqué que les deux types d'addition peuvent se produire et dépendent de la concentration de chlorure[18] - [19]. Cependant, ces recherches sont aussi contestées car les alcools allyliques peuvent être sensibles à des réactions d'isomérisation. Ainsi, différents stéréoisomères peuvent être formés à partir de ces réactions et non du procédé Wacker.
En résumé, l'expérimentation semble soutenir que les additions syn se produisent à des concentrations faibles en chlorure (< 1 mol/L, en conditions industrielles), alors que les additions anti se produisent quand la concentration en chlorure est élevée (> 3 mol/L). Ce phénomène est probablement dû à des ions chlorure capable de saturer le catalyseur et d'inhiber le mécanisme. Cependant, la raison de cette modification de mécanisme est encore inconnue.
Le rôle du chlorure de cuivre vient compliquer encore plus le procédé Wacker. La plupart des théories indiqueraient que le cuivre n'a pas de rôle dans l'oxydation de l'oléfine. Pourtant, des expériences menées par Stangl et Jira[20] ont montré que la formation de la chlorohydrine dépendait des concentrations en chlorure de cuivre. Les travaux de Hosokawa et de ses collaborateurs[21] ont donné un produit cristallisé contenant du chlorure de cuivre. Cela indique qu'il peut participer à l'oxydation de l'oléfine. Enfin, une étude ab initio de Comas-Vives et al.[22] montre que sans cuivre comme co-catalyseur, le mécanisme préfère une addition anti. Ce mécanisme a été confirmé plus tard par les expériences sans cuivre d'Anderson et Sigman[23]. Bien que ces travaux compliquent le mécanisme du procédé Wacker, il faut sans doute en déduire que cette réaction chimique peut être sensible aux conditions de réaction, et que plusieurs mécanismes réactionnels peuvent intervenir.
Une autre étape clé dans le procédé Wacker est la migration de l'hydrogène de l'oxygène au chlore et la formation de la double liaison C=O. On pense que cette étape passe par le biais de l'élimination d'un β-hydrure avec un état de transition cyclique à quatre chaînons :
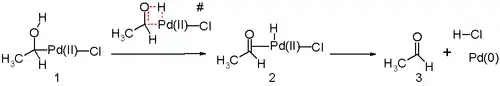
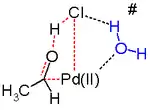
Des études in silico[24] - [25] affirment que l'état de transition de cette étape de la réaction est défavorable et qu'une élimination réductrice est en jeu. Cette étape alternative de la réaction est sans doute assistée par la molécule d'eau dans la solution qui agit comme un catalyseur.
Procédé industriel
Deux procédés sont commercialisés pour la production d'éthanal : il existe un processus à une étape et à deux étapes.
Procédé à une étape
L'éthylène et l'oxygène sont passés en co-courant dans une tour de réaction, à environ 130 °C et 400 kPa[26]. Le catalyseur est une solution aqueuse de PdCl2 et CuCl2. L'éthanal est purifié par une extraction suivie par une distillation fractionnée. L'extraction avec de l'eau supprime les composés les plus légers, ceux ayant un point d'ébullition plus faible que l'éthanal (chlorométhane, chloroéthane, et le dioxyde de carbone), qui arrivent dans la partie supérieure de la colonne, tandis que l'eau et les sous-produits avec des températures d'ébullition plus élevées (acide acétique, crotonaldéhyde ou éthanal chloré) sont retirés avec de l'éthanal en bas[26]. En raison de la nature corrosive du catalyseur, le réacteur est doublé avec de la céramique à l'épreuve de l'acide et le tube est fait en titane.
Procédé à deux étapes
Dans un processus à deux étapes, la réaction et l'oxydation sont effectuées séparément dans des réacteurs tubulaires. Contrairement au procédé à une étape, l'air peut être utilisé à la place de l'oxygène. L'éthylène est passé à travers le réacteur avec le catalyseur à 105-110 °C et 900-1 000 kPa[26]. Le catalyseur qui contient l'éthanal est séparé par une distillation flash. Ce mélange est oxydé dans le réacteur d'oxydation à 1 000 kPa en utilisant l'air comme oxydant. La solution de catalyseur oxydé est séparé et ernvoyé vers le réacteur. L'oxygène de l'air est entièrement utilisé et l'air appauvrie est distribué sous gaz inerte. Le mélange éthanal/vapeur d'eau obtient un ratio de 60 % à 90 % en éthanal grâce à la chaleur de la réaction. L'eau rejetée est retourné à la tour de distillation flash pour maintenir la concentration en catalyseur. Une distillation en deux étapes de l'éthanal brut vient ensuite. Durant la première phase, les substances ayant un point d'ébullition bas, tels que le chlorométhane, chloroéthane et de dioxyde de carbone, sont séparés. Durant la deuxième phase, l'eau et les sous-produits à point d'ébullition haut, tels que l'acide acétique et l'acétaldéhyde chloré, sont supprimés et l'éthanal est ainsi purifié[26]. En raison de la nature corrosive du catalyseur, les équipements qui sont en contact avec lui sont bordés de titane.
Dans les deux procédés, le rendement de la réaction est d'environ 95 %[26] et les coûts de production sont pratiquement les mêmes. Le gain de coût de l'utilisation de des gaz dilués dans la méthode à deux étapes est équilibré par des coûts d'investissement plus élevés. Les deux méthodes donnent des hydrocarbures chlorés, de l'éthanal chloré, et de l'acide acétique comme sous-produits. Généralement, le choix de la méthode dépend des matières premières et de l'énergie ainsi que par la disponibilité de l'oxygène à un prix raisonnable. En général, 100 parties d'éthylène donnent :
- 95 parties d'éthanal
- 1,9 partie d'aldéhydes chlorés
- 1,1 partie d'éthylène non transformé
- 0,8 partie de dioxyde de carbone
- 0,7 partie d'acide acétique
- 0,1 partie de chlorométhane
- 0,1 partie de chlorure d'éthyle
- 0,3 partie d'éthane, méthane, crotonaldéhyde
et d'autres sous-produits mineurs.
 Un diagramme montrant le diagramme de flux de processus pour le procédé Wacker à deux étapes de fabrication de l'acétaldéhyde.
Un diagramme montrant le diagramme de flux de processus pour le procédé Wacker à deux étapes de fabrication de l'acétaldéhyde. Un diagramme montrant le diagramme de flux de processus pour le procédé Wacker à une étape de fabrication de l'acétaldéhyde.
Un diagramme montrant le diagramme de flux de processus pour le procédé Wacker à une étape de fabrication de l'acétaldéhyde.
Oxydation Wacker–Tsuji
L'oxydation Wacker–Tsuji est la version de laboratoire de la réaction. Par exemple on peut y convertir du 1-décène en 2-décanone avec du chlorure de palladium(II) et du chlorure de cuivre(I) dans un mélange de solvant eau/diméthylformamide, en présence d'air[27] :
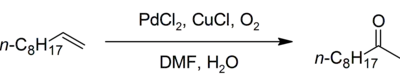
Certaines des réactions chimiques sur des oxydations et des aminations similiaires à celles de Wacker ont été examinées par Stahl et son équipe[28].
Références
- Traduit en partie de de:Wacker-Verfahren.
- Elschenbroich, C., Organometallics, 2006, Wiley-VCH, Weinheim.
- Acetaldehyde from Ethylene — A Retrospective on the Discovery of the Wacker Process Reinhard Jira, Angew.
- J. Smidt, W. Hafner, J. Sedlmeier, R. Jira, R. Rottinger (Cons. f.elektrochem.)
- F. C. Phillips, Am. Chem.
- F. C. Phillips, Z. Anorg.
- J. Smidt, W. Hafner, R. Jira, J. Sedlmeier, R. Sieber, R. Rüttinger et H. Kojer, Angew.
- W. Hafner, R. Jira, J. Sedlmeier et J. Smidt, Chem.
- J. Smidt, W. Hafner, R. Jira, R. Sieber, J. Sedlmeier et A. Sabel, Angew.
- Henry, Patrick M. In Handbook of Organopalladium Chemistry for Organic Synthesis, Negishi, E., Éd., Wiley & Sons, New York, 2002, p. 2119.
- J.A. Keith et P.M. Henry, Angew.
- James, D.E., Stille, J.K., J. Organomet.
- Stille, J.K., Divakarumi, R.J., J. Organomet.
- James, D.E., Hines, L.F., Stille, J.K., J. Am. Chem.
- Bäckvall, J.E., Akermark, B., Ljunggren, S.O., J. Am. Chem.
- Zaw, K., Lautens, M. et Henry P.M., Organometallics, 1985, 4, 1286–1296
- Wan W.K., Zaw K. et Henry P.M., Organometallics, 1988, 7, 1677–1683
- Francis, J.W., Henry, P.M. Organometallics, 1991, 10, 3498, DOI 10.1021/om00056a019
- Francis, J.W., Henry, P.M., Organometallics, 1992, 11, 2832, DOI 10.1021/om00044a024
- H. Stangl et R. Jira, Tetrahedron Lett., 1970, 11, 3589–3592
- T. Hosokawa, T. Nomura, S.-I. Murahashi, J. Organomet.
- Comas-Vives, A., Stirling, A., Ujaque, G., Lledós, A., Chem.
- Anderson, B.J., Keith, J.A. et Sigman, M.S., J. Am. Chem.
- J. A. Keith, J. Oxgaard et W. A. Goddard, III J. Am. Chem.
- H. E. Hosseini, S. A. Beyramabadi, A. Morsali et M. R. Housaindokht, J. Mol.
- Ullmann's Encyclopedia of Industrial Chemistry, Weinheim, Wiley-VCH, (DOI 10.1002/14356007.a01_031.pub2), « Acetaldehyde »
- Jiro Tsuji, Hideo Nagashima et Hisao Nemoto, General Synthetic Method for the preparation of Methyl Ketones from Terminal Olefins: 2-Decanone, Org. Synth., coll. « vol. 7 », , p. 137
- McDonald, R.I., Liu, G., Stahl, S.S., « Palladium(II)-Catalyzed Alkene Functionalization via Nucleopalladation: Stereochemical Pathways and Enantioselective Catalytic Applications », Chem. Rev., vol. 111, no 4, , p. 2981–3019 (DOI 10.1021/cr100371y)
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Wacker process » (voir la liste des auteurs).