En thermodynamique, une équation d'état cubique est une équation d'état d'un fluide pouvant s'écrire sous la forme d'un polynôme de degré trois en fonction du volume.
Elles ont une forme générale de polynôme du troisième degré en  , le facteur de compressibilité :
, le facteur de compressibilité :
Équation d'état cubique : 
avec  des paramètres qui ne dépendent que de la pression, de la température et de la composition du mélange, le volume n'apparaissant que dans le facteur de compressibilité. Sous cette forme, ces équations sont facilement utilisables en appliquant la méthode de Cardan pour trouver le volume d'un fluide.
des paramètres qui ne dépendent que de la pression, de la température et de la composition du mélange, le volume n'apparaissant que dans le facteur de compressibilité. Sous cette forme, ces équations sont facilement utilisables en appliquant la méthode de Cardan pour trouver le volume d'un fluide.
Équations diverses
Équation de van der Waals (1873)
Cette équation d'état, la première équation d'état cubique, a été proposée en 1873 par Johannes Diderik van der Waals[4]. Elle valut à celui-ci le prix Nobel de physique de 1910 « pour ses travaux sur l'équation d'état des gaz et des liquides »[5]. Elle est la première équation d'état qui, en tenant compte des interactions entre molécules via le paramètre  et de la taille de celles-ci via le paramètre
et de la taille de celles-ci via le paramètre  , représente la transition de phase liquide-gaz et le point critique. Elle se présente sous la forme :
, représente la transition de phase liquide-gaz et le point critique. Elle se présente sous la forme :
Équation de van der Waals : 
Elle a pour forme polynomiale :
Équation de van der Waals : 
Pour un corps pur, les paramètres (constant) et sont calculés à partir des pression et température critiques (mesurables expérimentalement) selon :
Pour un corps pur :


Le paramètre est généralement écrit sous la forme  . Pour l'équation de van der Waals,
. Pour l'équation de van der Waals,  et
et  .
.
Cette équation nécessite la connaissance de  et
et  .
.
Équation de Clausius (1880)
L'équation de van der Waals souffre de nombreux défauts, dont ceux de ne pas représenter correctement le volume critique et la capacité thermique isochore. Dès 1880 Rudolf Clausius, qui auparavant avait complété l'énoncé du deuxième principe de la thermodynamique (1850) et inventé le concept d'entropie (1865), proposa une amélioration de l'équation d'état de van der Waals[6] - [7] - [8]. Cette équation tente de corriger le défaut de l'équation de van der Waals au point critique en introduisant un terme correctif  et celui concernant la capacité thermique isochore en faisant dépendre le paramètre de la température. L'équation de Clausius prend la forme :
et celui concernant la capacité thermique isochore en faisant dépendre le paramètre de la température. L'équation de Clausius prend la forme :
Équation de Clausius : 
Elle a pour forme polynomiale :
Équation de Clausius : 
Pour un corps pur, les paramètres , et sont calculés à partir des coordonnées du point critique (mesurables expérimentalement) selon :
Pour un corps pur :



avec :

Cette équation nécessite la connaissance de , et  (ou
(ou  ).
).
Équation de Berthelot (1899)
Le paramètre de l'équation de Clausius étant petit devant , Daniel Berthelot propose en 1899 de considérer que  [9] - [10] - [11]. Son équation prend la forme :
[9] - [10] - [11]. Son équation prend la forme :
Équation de Berthelot : 
Elle a pour forme polynomiale :
Équation de Berthelot :
Pour un corps pur, les paramètres et sont calculés à partir des coordonnées du point critique (mesurables expérimentalement) selon :
Pour un corps pur :


avec :
Cette équation nécessite la connaissance de , et (ou ).
Équation de Redlich-Kwong (1949)
En 1949 Otto Redlich et Joseph Neng Shun Kwong proposent une nouvelle modification du terme attractif de l'équation de van der Waals[12] - [13] :
Équation de Redlich-Kwong : 
Elle a pour forme polynomiale :
Équation de Redlich-Kwong : 
Pour un corps pur, les paramètres et sont calculés à partir des coordonnées du point critique (mesurables expérimentalement) selon :
Pour un corps pur :
![{\displaystyle a={1 \over 9\left({\sqrt[{3}]{2}}-1\right)}{R^{2}{T_{\mathrm {c} }}^{2} \over P_{\mathrm {c} }}\cdot \alpha \!\left(T\right)=0,42748{R^{2}{T_{\mathrm {c} }}^{2} \over P_{\mathrm {c} }}\cdot \alpha \!\left(T\right)}](https://img.franco.wiki/i/68032ef3575ed6fb4165d9403728651114fb2b04.svg)
![{\displaystyle b={{\sqrt[{3}]{2}}-1 \over 3}{RT_{\mathrm {c} } \over P_{\mathrm {c} }}=0,08664{RT_{\mathrm {c} } \over P_{\mathrm {c} }}}](https://img.franco.wiki/i/7735532772d681c050634778c6a25afee9d96fde.svg)
avec :

Cette équation nécessite la connaissance de et .
Équation de Soave-Redlich-Kwong (1972)
En 1972 Giorgio Soave propose une modification de la fonction  de l'équation de Redlich-Kwong en y introduisant le facteur acentrique
de l'équation de Redlich-Kwong en y introduisant le facteur acentrique  [14]. Cette nouvelle équation est appelée équation de Soave-Redlich-Kwong ou de Redlich-Kwong-Soave. Elle est de la forme :
[14]. Cette nouvelle équation est appelée équation de Soave-Redlich-Kwong ou de Redlich-Kwong-Soave. Elle est de la forme :
Équation de Soave-Redlich-Kwong :
Elle a pour forme polynomiale :
Équation de Soave-Redlich-Kwong :
Pour un corps pur, les paramètres et sont calculés à partir des coordonnées du point critique (mesurables expérimentalement) selon :
Pour un corps pur :
avec :


Cette équation nécessite la connaissance de , et .
Équation de Peng-Robinson (1976-1978)
En 1976 Ding-Yu Peng et Donald Robinson proposent une nouvelle modification du terme attractif de l'équation de van der Waals[15]. Cette équation est de la forme :
Équation de Peng-Robinson : 
Elle a pour forme polynomiale :
Équation de Peng-Robinson : 
Pour un corps pur, les paramètres et sont calculés à partir des coordonnées du point critique (mesurables expérimentalement) selon :
Pour un corps pur :


avec :

Cette équation est appelée Peng-Robinson 76.
En 1978, Peng et Robinson modifient la fonction  selon[16] :
selon[16] :
 si
si 
 sinon
sinon
Cette équation est appelée Peng-Robinson 78.
Cette équation nécessite la connaissance de , et .
Équation de Patel-Teja (1982)
En 1982 Navin C. Patel et Amin S. Teja proposent une nouvelle modification du terme attractif de l'équation de van der Waals afin de mieux prendre en compte le facteur de compressibilité critique via un paramètre [17] - [18]. Cette équation est de la forme :
Équation de Patel-Teja : 
Elle a pour forme polynomiale :
Équation de Patel-Teja : 
Pour un corps pur, les paramètres , et sont calculés à partir des coordonnées du point critique (mesurables expérimentalement) selon :
Pour un corps pur :


avec :

Le facteur de compressibilité critique peut être la valeur expérimentale ou être calculé, pour les fluides apolaires, selon :

Le paramètre  est la plus petite racine réelle de l'équation :
est la plus petite racine réelle de l'équation :

Les paramètres  et
et  sont ensuite calculés selon :
sont ensuite calculés selon :


Cette équation est proche de celle de Soave-Redlich-Kwong si  et de celle de Peng-Robinson si
et de celle de Peng-Robinson si  .
.
Cette équation nécessite la connaissance de , , et (ou ).
Équation de Patel-Teja-Valderrama (1990)
En 1990, la complexité de la formulation de l'équation de Patel-Teja et sa limitation aux fluides apolaires conduisent José O. Valderrama à en proposer une forme généralisée, plus simple mathématiquement et applicable également aux fluides polaires[19]. Cette équation est de la forme :
Équation de Patel-Teja-Valderrama :
Elle a pour forme polynomiale :
Équation de Patel-Teja-Valderrama :
Pour un corps pur, les paramètres , et sont calculés à partir des coordonnées du point critique (mesurables expérimentalement) selon :
Pour un corps pur :
avec :

Les paramètres , et sont calculés selon :



Cette équation nécessite la connaissance de , , et (ou ).
Toutes les équations d'état cubiques présentées précédemment peuvent être représentées sous les deux formes généralisées suivantes :
Équation d'état cubique généralisée
(FG1)  (FG2)
(FG2) 
Les paramètres  ,
,  ,
,  et
et  sont des nombres réels adimensionnels. Ils sont liés par les relations :
sont des nombres réels adimensionnels. Ils sont liés par les relations :


et réciproquement :


Selon l'équation d'état et les règles de mélange appliquées, les paramètres , , et peuvent dépendre de la température et de la composition. Le tableau suivant donne l'expression de ces paramètres pour les différentes équations d'état vues précédemment.
Paramètres des équations d'état cubiques
| Équation d'état cubique |
|
|
|
|
van der Waals
Berthelot |
 |
|
|
|
| Clausius |
 |
 |
 |
|
Redlich-Kwong
Soave-Redlich-Kwong |
 |
|
|
 |
| Peng-Robinson |
 |
|
 |
 |
Patel-Teja
Patel-Teja-Valderrama |
 |
|
 |
 |
On pose également la forme généralisée :
Équation d'état cubique généralisée
(FG3) 
avec  et
et  .
.
Par définition, toutes les équations d'état cubiques peuvent se mettre sous la forme générale de polynôme du troisième degré en , le facteur de compressibilité :
Équation d'état cubique généralisée :
(FP)
avec :
Le volume molaire  n'apparait que dans le facteur de compressibilité .
n'apparait que dans le facteur de compressibilité .
Le tableau suivant donne la valeur de ces paramètres pour les différentes équations d'état vues précédemment.
Paramètres des équations d'état cubiques
| Équation d'état cubique |
 |
 |
 |
van der Waals
Berthelot |
 |
 |
 |
| Clausius |
 |
 |
 |
Redlich-Kwong
Soave-Redlich-Kwong |
|
 |
|
| Peng-Robinson |
 |
 |
 |
Patel-Teja
Patel-Teja-Valderrama |
 |
 |
 |
Règles de mélange
Les règles de mélange permettent l'application des équations d'état aux mélanges, c'est-à-dire de calculer les paramètres , et , notés respectivement  ,
,  et
et  , pour un mélange. Ces paramètres font intervenir la composition du mélange via les fractions molaires de ses constituants
, pour un mélange. Ces paramètres font intervenir la composition du mélange via les fractions molaires de ses constituants  et les paramètres
et les paramètres  ,
,  et
et  du corps
du corps  pur pour chacun des
pur pour chacun des  corps du mélange.
corps du mélange.
Règles classiques - van der Waals (1890)
Ces règles sont dues à van der Waals (1890[20]) pour l'application de son équation d'état aux mélanges. Ces règles sont appelées règles de mélange classiques :
Règles de mélange classiques :



avec  un paramètre d'interaction binaire entre le corps et le corps
un paramètre d'interaction binaire entre le corps et le corps  , déterminé expérimentalement, avec
, déterminé expérimentalement, avec  et
et  .
.
- Variantes
Il existe des variantes de ces règles, l'une des plus simples consistant à calculer le covolume selon :

avec  un paramètre d'interaction binaire entre le corps et le corps , déterminé expérimentalement, avec
un paramètre d'interaction binaire entre le corps et le corps , déterminé expérimentalement, avec  et
et  . Si tous les sont nuls, cette règle revient à la règle classique.
. Si tous les sont nuls, cette règle revient à la règle classique.
Une autre variante consiste à calculer le covolume selon une combinaison des diamètres des particules :

ou de la surface de leur section :

Règles de Huron-Vidal (1979)
En 1979 Huron et Vidal[21] proposent des règles dans lesquelles le covolume du mélange est calculé selon la règle de van der Waals :
Mais le terme de cohésion, pour une équation d'état sans paramètre (van der Waals, Berthelot, Redlich-Kwong, Preng-Robinson...), est calculé selon :

avec :
 une constante propre à chaque équation d'état :
une constante propre à chaque équation d'état :  avec et les paramètres de la forme généralisée (FG2) de l'équation d'état cubique ; si
avec et les paramètres de la forme généralisée (FG2) de l'équation d'état cubique ; si  (y compris si
(y compris si  ), alors
), alors  ;
; un modèle d'enthalpie libre molaire d'excès extérieur à l'équation d'état (comme les modèles de Margules, Van Laar (en), Wilson, NRTL (en), UNIQUAC, UNIFAC ou COSMOSPACE).
un modèle d'enthalpie libre molaire d'excès extérieur à l'équation d'état (comme les modèles de Margules, Van Laar (en), Wilson, NRTL (en), UNIQUAC, UNIFAC ou COSMOSPACE).
Pour une équation d'état avec un paramètre (Clausius, Patel-Teja...), il faut ajouter la règle de mélange :
et modifier la règle de mélange du paramètre de cohésion :

avec :
 la constante calculée pour le mélange ;
la constante calculée pour le mélange ; la constante calculée pour le corps pur.
la constante calculée pour le corps pur.
Le principal défaut de ces règles est qu'elles prennent pour référence un état à pression infinie, or les modèles d'enthalpie libre molaire d'excès sont développés pour des pressions de l'ordre de la pression atmosphérique. Afin de corriger ce défaut, ces règles ont fait l'objet de deux modifications importantes par Michelsen en 1990[22] et par Dahl et Michelsen également en 1990[23].
Démonstration
L'enthalpie libre molaire idéale d'un mélange liquide s'écrit :
![{\displaystyle {\bar {G}}_{m}^{\text{l,id}}=\sum _{i=1}^{N}x_{i}\left[{\bar {G}}_{i}^{\text{l,*}}+RT\,\ln x_{i}\right]}](https://img.franco.wiki/i/05bd3f22c7a1027ad3624cf1b4c4faf615fa09e2.svg)
avec :
 l'enthalpie libre molaire du mélange liquide idéal aux mêmes pression, température et composition que le mélange réel ;
l'enthalpie libre molaire du mélange liquide idéal aux mêmes pression, température et composition que le mélange réel ; l'enthalpie libre molaire du corps pur à l'état liquide aux mêmes pression et température que le mélange réel ;
l'enthalpie libre molaire du corps pur à l'état liquide aux mêmes pression et température que le mélange réel ;- la fraction molaire du corps .
L'enthalpie libre molaire d'excès d'une phase liquide s'écrit :
![{\displaystyle {\bar {G}}_{m}^{\text{E}}={\bar {G}}_{m}^{\text{l}}-{\bar {G}}_{m}^{\text{l,id}}={\bar {G}}_{m}^{\text{l}}-\sum _{i=1}^{N}x_{i}\left[{\bar {G}}_{i}^{\text{l,*}}+RT\,\ln x_{i}\right]}](https://img.franco.wiki/i/32562c0bf270bf59ffeeb54f28193fe882450fad.svg)
avec :
 l'enthalpie libre molaire d'excès ;
l'enthalpie libre molaire d'excès ; l'enthalpie libre molaire du mélange liquide réel.
l'enthalpie libre molaire du mélange liquide réel.
L'enthalpie libre molaire du mélange de gaz parfaits correspondant au mélange liquide réel s'écrit :
![{\displaystyle {\bar {G}}_{m}^{\bullet }=\sum _{i=1}^{N}x_{i}\left[{\bar {G}}_{i}^{\bullet ,*}+RT\,\ln x_{i}\right]}](https://img.franco.wiki/i/2f23b19eb0e51cd55952ed09c61f2ddd88b0fb59.svg)
avec :
 l'enthalpie libre molaire du mélange de gaz parfaits aux mêmes pression, température et composition que le mélange réel ;
l'enthalpie libre molaire du mélange de gaz parfaits aux mêmes pression, température et composition que le mélange réel ; l'enthalpie libre molaire du corps à l'état de gaz parfait pur aux mêmes pression et température que le mélange réel.
l'enthalpie libre molaire du corps à l'état de gaz parfait pur aux mêmes pression et température que le mélange réel.
On introduit l'enthalpie libre molaire du mélange de gaz parfaits dans l'enthalpie libre molaire d'excès :
![{\displaystyle {\bar {G}}_{m}^{\text{E}}={\bar {G}}_{m}^{\text{l}}-{\bar {G}}_{m}^{\bullet }-\sum _{i=1}^{N}x_{i}\left[{\bar {G}}_{i}^{\text{l,*}}+RT\,\ln x_{i}\right]+{\bar {G}}_{m}^{\bullet }=\left[{\bar {G}}_{m}^{\text{l}}-{\bar {G}}_{m}^{\bullet }\right]-\sum _{i=1}^{N}x_{i}\left[{\bar {G}}_{i}^{\text{l,*}}-{\bar {G}}_{i}^{\bullet ,*}\right]}](https://img.franco.wiki/i/fdea1d069e49cf882cae81c4cf49e9afc093b54e.svg)
On reconnait les enthalpies libres molaires résiduelles du mélange et de chacun des corps purs :


avec :
 l'enthalpie libre molaire résiduelle du mélange ;
l'enthalpie libre molaire résiduelle du mélange ; l'enthalpie libre molaire résiduelle du corps pur.
l'enthalpie libre molaire résiduelle du corps pur.
On a donc :

L'enthalpie libre molaire d'un mélange liquide s'écrit, avec la forme généralisée (FG2) des équations d'état cubiques :

L'enthalpie libre molaire résiduelle d'un liquide pur s'écrit, avec la forme généralisée (FG2) des équations d'état cubiques :

En remplaçant les enthalpies libres résiduelles par leur expression et en réarrangeant on a :
![{\displaystyle {\bar {G}}_{m}^{\text{E}}={a_{m} \over b_{m}}{\mathcal {F}}_{m}\!\left({\bar {V}}_{m},n\right)-\sum _{i=1}^{N}x_{i}{a_{i} \over b_{i}}{\mathcal {F}}_{i}\!\left({\bar {V}}_{i}^{\text{l,*}}\right)+P\left[{\bar {V}}_{m}^{\text{l}}-\sum _{i=1}^{N}x_{i}{\bar {V}}_{i}^{\text{l,*}}\right]+\sum _{i=1}^{N}x_{i}RT\,\ln \!\left({{\bar {V}}_{i}^{\text{l,*}}-b_{i} \over {\bar {V}}_{m}^{\text{l}}-b_{m}}\right)}](https://img.franco.wiki/i/f6addc075c5b751d38e762e24a19336cd06b7598.svg)
On pose la règle de mélange identique à celle de van der Waals pour le covolume :
on a :
![{\displaystyle {\bar {G}}_{m}^{\text{E}}={a_{m} \over b_{m}}{\mathcal {F}}_{m}\!\left({\bar {V}}_{m},n\right)-\sum _{i=1}^{N}x_{i}{a_{i} \over b_{i}}{\mathcal {F}}_{i}\!\left({\bar {V}}_{i}^{\text{l,*}}\right)+P\left[\left({\bar {V}}_{m}^{\text{l}}-b_{m}\right)-\sum _{i=1}^{N}x_{i}\left({\bar {V}}_{i}^{\text{l,*}}-b_{i}\right)\right]+\sum _{i=1}^{N}x_{i}RT\,\ln \!\left({{\bar {V}}_{i}^{\text{l,*}}-b_{i} \over {\bar {V}}_{m}^{\text{l}}-b_{m}}\right)}](https://img.franco.wiki/i/65e0a9172ba8624b3c46312b13a47b18892d41d2.svg)
Pour le mélange et le corps pur :




D'où, à pression infinie :


Si  , c'est-à-dire si les paramètres et de la forme généralisée (FG2) des équations d'état cubiques sont les mêmes pour un corps pur et pour un mélange, ce qui exclut les équations possédant un paramètre (Clausius, Patel-Teja...) :
, c'est-à-dire si les paramètres et de la forme généralisée (FG2) des équations d'état cubiques sont les mêmes pour un corps pur et pour un mélange, ce qui exclut les équations possédant un paramètre (Clausius, Patel-Teja...) :

Règles de Wong-Sandler (1992)
En 1992 Wong et Sandler[24] proposent, pour une équation d'état sans paramètre (van der Waals, Berthelot, Redlich-Kwong, Preng-Robinson...), de modifier la règle de mélange du covolume selon :

avec :
- une constante propre à chaque équation d'état : avec et les paramètres de la forme généralisée (FG2) de l'équation d'état cubique ; si (y compris si ), alors ;
- un modèle d'enthalpie libre molaire d'excès extérieur à l'équation d'état (comme les modèles de Margules, Van Laar (en), Wilson, NRTL (en), UNIQUAC, UNIFAC ou COSMOSPACE) ;
![{\displaystyle \left(b-{a \over RT}\right)_{i,j}={1 \over 2}\left[\left(b_{i}-{a_{i} \over RT}\right)+\left(b_{j}-{a_{j} \over RT}\right)\right]\left(1-k_{i,j}\right)}](https://img.franco.wiki/i/13e6fceda56c666eb52d5dcc2b29178da38f3a7c.svg) ; étant un paramètre d'interaction binaire entre le corps et le corps , déterminé expérimentalement, avec et .
; étant un paramètre d'interaction binaire entre le corps et le corps , déterminé expérimentalement, avec et .
Le terme de cohésion est calculé selon :

Pour une équation d'état avec un paramètre (Clausius, Patel-Teja...), il faut ajouter la règle de mélange :
et modifier la règle de mélange du covolume :

avec :
- la constante calculée pour le mélange ;
- la constante calculée pour le corps pur.
Contrairement aux règles de van der Waals et de Huron-Vidal, les règles de Wong-Sandler impliquent une dépendance du paramètre à la température, même si les paramètres des corps purs sont des constantes.
Démonstration
L'énergie libre molaire idéale d'un mélange liquide s'écrit :
![{\displaystyle {\bar {F}}_{m}^{\text{l,id}}=\sum _{i=1}^{N}x_{i}\left[{\bar {F}}_{i}^{\text{l,*}}+RT\,\ln x_{i}\right]}](https://img.franco.wiki/i/52b363820c3a785ab5fc3be5bf354ceae4db7901.svg)
avec :
 l'énergie libre molaire du mélange liquide idéal aux mêmes pression, température et composition que le mélange réel ;
l'énergie libre molaire du mélange liquide idéal aux mêmes pression, température et composition que le mélange réel ; l'énergie libre molaire du corps pur à l'état liquide aux mêmes pression et température que le mélange réel ;
l'énergie libre molaire du corps pur à l'état liquide aux mêmes pression et température que le mélange réel ;- la fraction molaire du corps .
L'énergie libre molaire d'excès d'une phase liquide s'écrit :
![{\displaystyle {\bar {F}}_{m}^{\text{E}}={\bar {F}}_{m}^{\text{l}}-{\bar {F}}_{m}^{\text{l,id}}={\bar {F}}_{m}^{\text{l}}-\sum _{i=1}^{N}x_{i}\left[{\bar {F}}_{i}^{\text{l,*}}+RT\,\ln x_{i}\right]}](https://img.franco.wiki/i/627b6b85602d1cb628c71a957137bf3f47bbe1e0.svg)
avec :
 l'énergie libre molaire d'excès ;
l'énergie libre molaire d'excès ; l'énergie libre molaire du mélange liquide réel.
l'énergie libre molaire du mélange liquide réel.
L'énergie libre molaire du mélange de gaz parfaits correspondant au mélange liquide réel s'écrit :
![{\displaystyle {\bar {F}}_{m}^{\bullet }=\sum _{i=1}^{N}x_{i}\left[{\bar {F}}_{i}^{\bullet ,*}+RT\,\ln x_{i}\right]}](https://img.franco.wiki/i/462977e7e2822dffd41db5250ec93767101c86cb.svg)
avec :
 l'énergie libre molaire du mélange de gaz parfaits aux mêmes pression, température et composition que le mélange réel ;
l'énergie libre molaire du mélange de gaz parfaits aux mêmes pression, température et composition que le mélange réel ; l'énergie libre molaire du corps à l'état de gaz parfait pur aux mêmes pression et température que le mélange réel.
l'énergie libre molaire du corps à l'état de gaz parfait pur aux mêmes pression et température que le mélange réel.
On introduit l'énergie libre molaire du mélange de gaz parfaits dans l'énergie libre molaire d'excès :
![{\displaystyle {\bar {F}}_{m}^{\text{E}}={\bar {F}}_{m}^{\text{l}}-{\bar {F}}_{m}^{\bullet }-\sum _{i=1}^{N}x_{i}\left[{\bar {F}}_{i}^{\text{l,*}}+RT\,\ln x_{i}\right]+{\bar {F}}_{m}^{\bullet }=\left[{\bar {F}}_{m}^{\text{l}}-{\bar {F}}_{m}^{\bullet }\right]-\sum _{i=1}^{N}x_{i}\left[{\bar {F}}_{i}^{\text{l,*}}-{\bar {F}}_{i}^{\bullet ,*}\right]}](https://img.franco.wiki/i/83dfe638c0e595b9cc5418ef533dcb13aaa1352a.svg)
On reconnait les énergies libres molaires résiduelles du mélange et de chacun des corps purs :


avec :
 l'énergie libre molaire résiduelle du mélange ;
l'énergie libre molaire résiduelle du mélange ; l'énergie libre molaire résiduelle du corps pur.
l'énergie libre molaire résiduelle du corps pur.
On a donc :

L'énergie libre molaire d'un mélange liquide s'écrit, avec la forme généralisée (FG2) des équations d'état cubiques :

L'énergie libre molaire résiduelle d'un liquide pur s'écrit, avec la forme généralisée (FG2) des équations d'état cubiques :

En remplaçant les énergies libres résiduelles par leur expression et en réarrangeant on a :

Pour le mélange et le corps pur :
avec :
- la constante calculée pour le mélange ;
- la constante calculée pour le corps pur.
D'où, à pression infinie :

Wong et Sandler posent que l'énergie libre est peu sensible à la pression et qu'à basse pression elle est quasi égale à l'enthalpie libre ( , or
, or  ), d'où :
), d'où :

en conséquence :

Si , c'est-à-dire si les paramètres et de la forme généralisée (FG2) des équations d'état cubiques sont les mêmes pour un corps pur et pour un mélange, ce qui exclut les équations possédant un paramètre (Clausius, Patel-Teja...) :

Cette expression revient à celle de Huron-Vidal, mais il n'a pas été nécessaire d'imposer une règle de mélange sur . Pour trouver une deuxième règle de mélange, Wong et Sandler partent du principe que le second coefficient du viriel d'un mélange doit être une forme quadratique de la composition (fractions molaires). En multipliant par  la forme généralisée (FG2) des équations d'état cubiques, on obtient :
la forme généralisée (FG2) des équations d'état cubiques, on obtient :
![{\displaystyle Z={{\bar {V}} \over {\bar {V}}-b}-{{\bar {V}}{a \over RT} \over \left({\bar {V}}-br_{1}\right)\left({\bar {V}}-br_{2}\right)}={{\bar {V}} \over {\bar {V}}-b}-{{\bar {V}}{a \over RT} \over b\left(r_{1}-r_{2}\right)}\left[{1 \over {\bar {V}}-br_{1}}-{1 \over {\bar {V}}-br_{2}}\right]}](https://img.franco.wiki/i/036b8fd7d478d6ebabe76d11265539361430a33d.svg)
![{\displaystyle Z={1 \over 1-{b \over {\bar {V}}}}-{{a \over RT} \over b\left(r_{1}-r_{2}\right)}\left[{1 \over 1-{b \over {\bar {V}}}r_{1}}-{1 \over 1-{b \over {\bar {V}}}r_{2}}\right]}](https://img.franco.wiki/i/efa2d60749242377d0cb5d89209f5c24dd151a46.svg)
Pour de grands volumes molaires,  , soit
, soit  , on a par développement limité :
, on a par développement limité :



On obtient par conséquent un développement sous forme d'équation du viriel :
![{\displaystyle Z=1+{b \over {\bar {V}}}+{b^{2} \over {\bar {V}}^{2}}+{b^{3} \over {\bar {V}}^{3}}-{{a \over RT} \over b\left(r_{1}-r_{2}\right)}\left[{br_{1} \over {\bar {V}}}+{b^{2}{r_{1}}^{2} \over {\bar {V}}^{2}}+{b^{3}{r_{1}}^{3} \over {\bar {V}}^{3}}-{br_{2} \over {\bar {V}}}-{b^{2}{r_{2}}^{2} \over {\bar {V}}^{2}}-{b^{3}{r_{2}}^{3} \over {\bar {V}}^{3}}\right]+\cdots }](https://img.franco.wiki/i/ea44aa94266cd8619a6a0af65366ccbefc79f952.svg)
![{\displaystyle =1+{b \over {\bar {V}}}+{b^{2} \over {\bar {V}}^{2}}+{b^{3} \over {\bar {V}}^{3}}-{{a \over RT} \over b\left(r_{1}-r_{2}\right)}\left[{br_{1}-br_{2} \over {\bar {V}}}+{b^{2}{r_{1}}^{2}-b^{2}{r_{2}}^{2} \over {\bar {V}}^{2}}+{b^{3}{r_{1}}^{3}-b^{3}{r_{2}}^{3} \over {\bar {V}}^{3}}\right]+\cdots }](https://img.franco.wiki/i/25e0761acd693fc938d35d10b6908206cd1a5d16.svg)
En tronquant à l'ordre 2 :
![{\displaystyle Z=1+\left[b-{a \over RT}\right]{1 \over {\bar {V}}}+\cdots }](https://img.franco.wiki/i/1f6034ea47b376d204da9e1914cbba592e946906.svg)
Le second coefficient du viriel vaut :  . La thermodynamique statique montre que le second coefficient du viriel d'un mélange est lié aux fractions molaires par une relation quadratique :
. La thermodynamique statique montre que le second coefficient du viriel d'un mélange est lié aux fractions molaires par une relation quadratique :

On pose :
![{\displaystyle B_{i,j}={1 \over 2}\left[B_{i}+B_{j}\right]\left(1-k_{i,j}\right)}](https://img.franco.wiki/i/65ee2bbf1e2a2c42cb9c6ab5c5d29d51608049b4.svg)
avec :
 le second coefficient du viriel du corps pur ;
le second coefficient du viriel du corps pur ;- un paramètre d'interaction binaire entre le corps et le corps , tel que et .
On a en conséquence :
Résolution d'une équation d'état cubique
Discriminant r
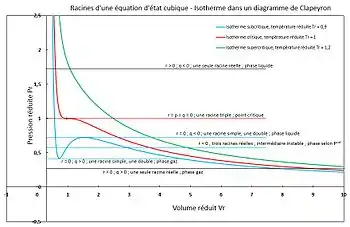
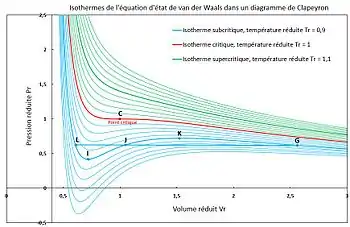
Figure 2 - Racines d'une équation d'état cubique. Signification en fonction de la résolution par la méthode de Cardan.
Sur une isotherme (à température donnée), pour une pression donnée, il peut correspondre une ou plusieurs racines (plusieurs volumes molaires) qui peuvent être attribuées selon le cas à une phase gaz ou une phase liquide.
Soit une équation d'état cubique mise sous sa forme polynomiale généralisée (FP) :
Équation d'état cubique :
Le volume molaire n'apparait que dans le facteur de compressibilité . Connaissant  ,
,  et la composition, le volume molaire du mélange est calculé à partir des racines du polynôme en :
et la composition, le volume molaire du mélange est calculé à partir des racines du polynôme en :

Les racines du polynôme peuvent être trouvées à l'aide de la méthode de Cardan. On pose :



 est un discriminant qui permet de distinguer trois cas (voir figure 2).
est un discriminant qui permet de distinguer trois cas (voir figure 2).
Cas r > 0
Si  le polynôme admet une racine réelle et deux complexes, seule la racine réelle nous intéresse :
le polynôme admet une racine réelle et deux complexes, seule la racine réelle nous intéresse :
![{\displaystyle Z_{0}={\sqrt[{3}]{{-q \over 2}+{\sqrt {r}}}}+{\sqrt[{3}]{{-q \over 2}-{\sqrt {r}}}}-{c_{2} \over 3}}](https://img.franco.wiki/i/8a18d899817a8d4e81d31f9503859e2d2e353846.svg)
On distingue deux cas :
- si
 le mélange est liquide,
le mélange est liquide,
- si
 le mélange est gazeux.
le mélange est gazeux.
Quelle que soit la température (critique, subcritique ou supercritique), les isothermes possèdent des branches correspondant à . Les isothermes supercritiques ne rencontrent que ce cas.
Cas r = 0
Si  le polynôme admet trois racines réelles dont une racine multiple :
le polynôme admet trois racines réelles dont une racine multiple :
- si
 alors la racine est triple et
alors la racine est triple et  , il s'agit du point critique, seule l'isotherme critique rencontre donc ce cas,
, il s'agit du point critique, seule l'isotherme critique rencontre donc ce cas,
- sinon, le polynôme admet une racine simple et une double :
racine simple : ![{\displaystyle Z_{0}=2\cdot {\sqrt[{3}]{-q \over 2}}-{c_{2} \over 3}}](https://img.franco.wiki/i/02e9748a940b3e2ada40378705da77fdbcb6ece8.svg) racine double :
racine double : ![{\displaystyle Z_{1}=Z_{2}=-{\sqrt[{3}]{-q \over 2}}-{c_{2} \over 3}}](https://img.franco.wiki/i/72d3c2b80f83bfb7d0e146e0924daa2d6b9b02cb.svg)
- On distingue deux cas :
- si la racine simple correspond à un liquide, la double à un gaz (point K de la figure 1),
- si la racine simple correspond à un gaz, la double à un liquide (point I de la figure 1).
- Dans les deux cas, la racine double correspond à une phase instable et doit être écartée. Seule la phase correspondant à la racine simple est stable et doit être retenue. Seules les isothermes subcritiques rencontrent le cas avec
 .
.
Cas r < 0
Si  le polynôme admet trois racines réelles distinctes. On pose :
le polynôme admet trois racines réelles distinctes. On pose :

Avec ![{\displaystyle k\in \left[0,1,2\right]}](https://img.franco.wiki/i/e867d5227eb76857b19cd28c5c87edaac858accd.svg) , on calcule les trois racines selon :
, on calcule les trois racines selon :

La plus grande racine permet de calculer le volume molaire d'un gaz (point G de la figure 1). La plus petite racine permet de calculer le volume molaire d'un liquide (point L de la figure 1). La racine intermédiaire (point J de la figure 1) n'a pas de réalité physique, il s'agirait d'une phase dont le volume molaire augmenterait avec une augmentation de pression (une phase ayant une compressibilité négative), ce qui n'existe pas.
Dans le cas d'un corps pur, si la pression  la pression de vapeur saturante, alors seul le liquide est stable. Inversement, si
la pression de vapeur saturante, alors seul le liquide est stable. Inversement, si  , seul le gaz est stable. Si
, seul le gaz est stable. Si  il y a équilibre liquide-vapeur, les deux phases sont stables. Seules les isothermes subcritiques rencontrent le cas .
il y a équilibre liquide-vapeur, les deux phases sont stables. Seules les isothermes subcritiques rencontrent le cas .
Résolution par ordinateur
Lors d'une résolution par ordinateur, les erreurs d'arrondi rendent le cas improbable. De façon générale, si tend vers 0, c'est-à-dire lorsque  et
et  sont proches en valeur absolue, mais opposés par le signe, le calcul devient imprécis. Il est alors nécessaire d'affiner les résultats par une autre méthode, comme la méthode de Newton que les résultats de la méthode de Cardan serviraient à initialiser[25] - [26].
sont proches en valeur absolue, mais opposés par le signe, le calcul devient imprécis. Il est alors nécessaire d'affiner les résultats par une autre méthode, comme la méthode de Newton que les résultats de la méthode de Cardan serviraient à initialiser[25] - [26].
Grandeurs calculables avec les équations d'état cubiques
Pression de vapeur saturante
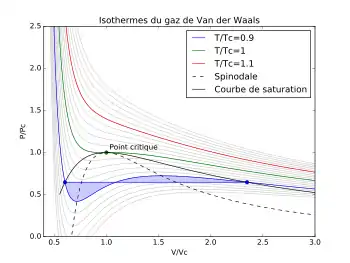
Figure 3 - Règle du palier de Maxwell appliquée à l'équation d'état de Van der Waals : la ligne horizontale est tracée de façon à couper la ligne isotherme théorique en délimitant deux surfaces d'aires égales.
Ce qui suit n'est applicable qu'à un corps pur. Le calcul des pression de bulle et de rosée d'un mélange est plus complexe et nécessite le calcul des coefficients de fugacité de chacun des composants du mélange.
Pour une température donnée inférieure à la température critique , pour des pressions inférieures à la pression critique , une équation d'état cubique peut produire trois valeurs du volume molaire d'un corps pur : le polynôme de degré trois en  peut en effet avoir trois racines réelles. La plus grande de ces racines permet le calcul d'un volume molaire attribuable à un gaz et noté
peut en effet avoir trois racines réelles. La plus grande de ces racines permet le calcul d'un volume molaire attribuable à un gaz et noté  . La plus petite de ces racines permet de calculer un volume molaire attribuable à un liquide et noté
. La plus petite de ces racines permet de calculer un volume molaire attribuable à un liquide et noté  . La racine intermédiaire n'a pas de sens physique, il s'agirait d'une phase dont le volume molaire augmenterait avec une augmentation de pression (une phase ayant une compressibilité négative), ce qui n'existe pas.
. La racine intermédiaire n'a pas de sens physique, il s'agirait d'une phase dont le volume molaire augmenterait avec une augmentation de pression (une phase ayant une compressibilité négative), ce qui n'existe pas.
Cependant, une équation d'état cubique suggère pour un corps pur la coexistence des deux phases liquide et gaz sur une certaine plage de pression pour une même température : selon les isothermes bleu turquoise de la figure 1, la coexistence des deux phases serait possible entre les pressions des points I et K. L'expérience toutefois montre que pour un corps pur cette coexistence, ou équilibre, à température donnée, n'a lieu qu'à une seule pression : la pression de vapeur saturante notée  . Les équations d'état cubiques sont donc incorrectes sur ce point et ne permettent pas le calcul direct des pressions de vapeur saturante, mais la règle du palier de Maxwell remédie à ce problème. Cette règle indique que le gaz et le liquide sont à l'équilibre si :
. Les équations d'état cubiques sont donc incorrectes sur ce point et ne permettent pas le calcul direct des pressions de vapeur saturante, mais la règle du palier de Maxwell remédie à ce problème. Cette règle indique que le gaz et le liquide sont à l'équilibre si :
Règle du palier de Maxwell : 
Ce qui revient à dire que les deux surfaces violettes représentées sur la figure 3 ci-dessus sont égales.
La forme généralisée (FG2) des équations d'état cubiques donne :

![{\displaystyle =RT\!\cdot \!{\Biggl [}\ln \!\left({\bar {V}}-b\right){\Biggr ]}_{{\bar {V}}^{\text{l}}}^{{\bar {V}}^{\text{g}}}-a\!\cdot \!\left[{{\mathcal {F}}\!\left({\bar {V}}\right) \over b}\right]_{{\bar {V}}^{\text{l}}}^{{\bar {V}}^{\text{g}}}=RT\!\cdot \!\ln \!\left({{\bar {V}}^{\text{g}}-b \over {\bar {V}}^{\text{l}}-b}\right)-{a \over b}\!\cdot \!\left({\mathcal {F}}\!\left({\bar {V}}^{\text{g}}\right)-{\mathcal {F}}\!\left({\bar {V}}^{\text{l}}\right)\right)}](https://img.franco.wiki/i/5828c8cda49783f6096bf7257578a2d5030899a0.svg)
On a donc, en appliquant la règle du palier de Maxwell à l'équation d'état cubique généralisée (FG2) :

La température étant donnée, on a un système de trois équations à trois inconnues (les volumes molaires et et la pression  ) :
) :



Le calcul de la pression de vapeur saturante d'un corps pur à température donnée  peut s'effectuer de façon itérative comme suit :
peut s'effectuer de façon itérative comme suit :
- fixer la pression
 ,
,
- calculer la ou les racines de la forme polynomiale cubique de l'équation d'état à et
 par la méthode de Cardan,
par la méthode de Cardan,
- si l'équation ne produit qu'une seule racine réelle, recommencer en 1,
- si l'équation produit trois racines réelles, calculer à partir de la plus grande racine et à partir de la plus petite racine,
- calculer
 ,
,
- si
 recommencer en 1,
recommencer en 1,
- sinon
 .
.
Une fois la pression de vapeur saturante connue, on peut écarter les phases métastables et instables lorsque l'équation d'état cubique produit trois racines réelles à la même température mais à d'autres pressions que la pression de vapeur saturante. En dessous de seule la phase gaz existe de façon stable : si l'équation d'état produit trois racines réelles seule celle attribuable à un gaz, la plus grande, doit être retenue. Au-dessus de seule la phase liquide est stable : si l'équation d'état produit trois racines réelles seule celle attribuable à un liquide, la plus petite, doit être retenue.
Pour un corps pur
Pour un corps pur (liquide ou gazeux) le coefficient de fugacité  calculé avec la forme généralisée (FG2) des équations d'état cubiques vaut :
calculé avec la forme généralisée (FG2) des équations d'état cubiques vaut :
Coefficient de fugacité d'un corps pur :

ou, sous forme adimensionnelle :

avec le coefficient de fugacité du corps pur.
À saturation, les coefficients de fugacité du corps pur en phases liquide et vapeur sont égaux. On note :
 le coefficient de fugacité du corps pur en phase gaz ;
le coefficient de fugacité du corps pur en phase gaz ; le coefficient de fugacité du corps pur en phase liquide ;
le coefficient de fugacité du corps pur en phase liquide ;- le volume molaire du corps pur en phase gaz ;
- le volume molaire du corps pur en phase liquide.
On a donc, à saturation :


En réarrangeant, on retrouve l'expression issue de la règle du palier de Maxwell appliquée à l'équation d'état cubique généralisée (FG2) :

Pour un corps dans un mélange
Dans ce qui suit un mélange (liquide ou gazeux) de corps est considéré. Les expressions données ci-dessous sont établies avec la forme généralisée (FG3) des équations d'état cubiques.
Le coefficient de fugacité de tout corps du mélange est calculé selon :
Coefficient de fugacité d'un corps en mélange :

![{\displaystyle -{a \over \left(b_{1}-b_{2}\right)}\,RT\,\left[{nb_{1,i}^{\prime }-nb_{2,i}^{\prime } \over b}{\mathcal {F}}\!\left({\bar {V}},z\right)+{nb_{1,i}^{\prime } \over {\bar {V}}+b_{1}}-{nb_{2,i}^{\prime } \over {\bar {V}}+b_{2}}\right]}](https://img.franco.wiki/i/8a4d7e8242150404fecbf7c5724364d5542912f9.svg)
ou, sous forme adimensionnelle :

![{\displaystyle -{A \over \left(B_{1}-B_{2}\right)}\left[{B_{1,i}^{\prime }-B_{2,i}^{\prime } \over B}{\mathcal {F}}\!\left(Z,z\right)+{B_{1,i}^{\prime } \over Z+B_{1}}-{B_{2,i}^{\prime } \over Z+B_{2}}\right]}](https://img.franco.wiki/i/e5c190f51df46352e0754f0d74e5021ecc17d821.svg)
avec :
 la dérivée partielle du terme de cohésion du mélange par rapport à la quantité
la dérivée partielle du terme de cohésion du mélange par rapport à la quantité  du corps , à température et quantités des autres corps constantes ;
du corps , à température et quantités des autres corps constantes ; le paramètre
le paramètre  normé ;
normé ; la dérivée partielle du covolume molaire du mélange par rapport à la quantité du corps , à température et quantités des autres corps constantes ;
la dérivée partielle du covolume molaire du mélange par rapport à la quantité du corps , à température et quantités des autres corps constantes ; le paramètre
le paramètre  normé ;
normé ; la dérivée partielle du paramètre
la dérivée partielle du paramètre  par rapport à la quantité du corps , à température et quantités des autres corps constantes ;
par rapport à la quantité du corps , à température et quantités des autres corps constantes ; le paramètre
le paramètre  normé ;
normé ; la dérivée partielle du paramètre
la dérivée partielle du paramètre  par rapport à la quantité du corps , à température et quantités des autres corps constantes ;
par rapport à la quantité du corps , à température et quantités des autres corps constantes ; le paramètre
le paramètre  normé ;
normé ; le coefficient de fugacité du corps en mélange.
le coefficient de fugacité du corps en mélange.
Pour un corps pur  , on vérifie que l'on retrouve l'expression de
, on vérifie que l'on retrouve l'expression de  .
.
Grandeurs résiduelles
Les grandeurs résiduelles  expriment l'écart entre les propriétés extensives d'un mélange réel
expriment l'écart entre les propriétés extensives d'un mélange réel  et celles d'un mélange de gaz parfaits
et celles d'un mélange de gaz parfaits  . Pour calculer une propriété d'un mélange réel (liquide ou gaz), il suffit donc de calculer la propriété correspondante d'un mélange de gaz parfaits et de lui ajouter la grandeur résiduelle appropriée, toutes ces propriétés étant calculées aux mêmes pression, température et composition que le mélange réel :
. Pour calculer une propriété d'un mélange réel (liquide ou gaz), il suffit donc de calculer la propriété correspondante d'un mélange de gaz parfaits et de lui ajouter la grandeur résiduelle appropriée, toutes ces propriétés étant calculées aux mêmes pression, température et composition que le mélange réel :
Grandeur résiduelle : 
Les grandeurs résiduelles sont calculées avec l'équation d'état cubique généralisée (FG2).
Les expressions données ci-dessous ne sont valables que si seul le paramètre dépend de la température.
Pour un corps pur comme pour un mélange (liquide ou gazeux), les grandeurs résiduelles valent (les expressions sont données à chaque fois sous forme dimensionnelle et sous forme adimensionnelle) :












avec :
 la dérivée partielle du terme de cohésion par rapport à la température à composition constante ;
la dérivée partielle du terme de cohésion par rapport à la température à composition constante ; le paramètre
le paramètre  normé.
normé.
Rappelons que par définition :
- pour un corps pur :
 ;
;
- pour un mélange :
 .
.
On remarquera également que pour un gaz parfait (corps pur ou mélange)  ,
,  et
et  : toutes les grandeurs résiduelles sont nulles comme il se doit.
: toutes les grandeurs résiduelles sont nulles comme il se doit.
Démonstration
Les grandeurs résiduelles sont calculées avec l'équation d'état cubique généralisée (FG2) :

- Calculs préalables
Calculons au préalable deux intégrales :
![{\displaystyle I_{1}=\int _{+\infty }^{V}\left[{1 \over V-nb}-{1 \over V}\right]\,\mathrm {d} V}](https://img.franco.wiki/i/4ac10ba31dfc47b988e4d482f8ca8b073342ada4.svg)

Pour la première :
![{\displaystyle I_{1}=\left[\,\ln \!\left(V-nb\right)-\ln V\,\right]_{+\infty }^{V}=\left[\,\ln \!\left({V-nb \over V}\right)\,\right]_{+\infty }^{V}}](https://img.franco.wiki/i/77f1592bc25f19ab74de9b7418ed06a725b32afa.svg)
En considérant que :

on a :
![{\displaystyle I_{1}=\int _{+\infty }^{V}\left[{1 \over V-nb}-{1 \over V}\right]\,\mathrm {d} V=\ln \!\left({V-nb \over V}\right)}](https://img.franco.wiki/i/07dcd99bdfa65f6ba1da0eedbd27fe98a96d1ac8.svg)
Pour la deuxième intégrale, si  on décompose en éléments simples :
on décompose en éléments simples :

On a donc :
![{\displaystyle I_{2}={1 \over nb\left(r_{1}-r_{2}\right)}\int _{+\infty }^{V}\left[{1 \over V-nbr_{1}}-{1 \over V-nbr_{2}}\right]\,\mathrm {d} V={1 \over nb\left(r_{1}-r_{2}\right)}\left[\,\ln \!\left(V-nbr_{1}\right)-\ln \!\left(V-nbr_{2}\right)\,\right]_{+\infty }^{V}={1 \over nb\left(r_{1}-r_{2}\right)}\left[\,\ln \!\left({V-nbr_{1} \over V-nbr_{2}}\right)\,\right]_{+\infty }^{V}}](https://img.franco.wiki/i/409d4f5218c8761ea9e5b0d71f420ebd0d9533b0.svg)
En considérant que :

on a :

Si , y compris si , on a :
![{\displaystyle I_{2}=\int _{+\infty }^{V}{1 \over \left(V-nbr_{1}\right)^{2}}\,\mathrm {d} V=\left[\,-{1 \over V-nbr_{1}}\,\right]_{+\infty }^{V}}](https://img.franco.wiki/i/12de1feac7832bf239cf6fdf5f7c0830bdf3c7e5.svg)
En considérant que :

on a :

On pose la fonction du volume  telle que :
telle que :
- si :

- si (y compris ) :

On a par conséquent :

- Calcul de l'énergie libre résiduelle molaire
L'énergie libre résiduelle molaire se calcule selon :
![{\displaystyle n{\bar {F}}^{\text{RES}}=-\int _{+\infty }^{V}\left[P-{nRT \over V}\right]\,\mathrm {d} V-nRT\,\ln Z}](https://img.franco.wiki/i/5a7ef978061301323934189ca997d87a921e5d37.svg)
On a donc, avec la forme généralisée (FG2) de l'équation d'état cubique :
![{\displaystyle n{\bar {F}}^{\text{RES}}=-\int _{+\infty }^{V}\left[{nRT \over V-nb}-{n^{2}a \over \left(V-nbr_{1}\right)\left(V-nbr_{2}\right)}-{nRT \over V}\right]\,\mathrm {d} V-nRT\,\ln Z}](https://img.franco.wiki/i/c7b472242e46595a6e8cd558ab2c0f3e2c83d9fd.svg)
![{\displaystyle =-nRT\int _{+\infty }^{V}\left[{1 \over V-nb}-{1 \over V}\right]\,\mathrm {d} V+n^{2}a\int _{+\infty }^{V}{1 \over \left(V-nbr_{1}\right)\left(V-nbr_{2}\right)}\,\mathrm {d} V-nRT\,\ln Z}](https://img.franco.wiki/i/54c4abac5d7b6c6a462ec58f56ce8f1e89d774d9.svg)

Avec par définition :

en réarrangeant on trouve :

- Calcul de l'entropie résiduelle molaire
L'entropie résiduelle molaire se calcule selon :
![{\displaystyle n{\bar {S}}^{\text{RES}}=\int _{+\infty }^{V}\left[\left({\partial P \over \partial T}\right)_{V,n}-{nR \over V}\right]\,\mathrm {d} V+nR\ln Z}](https://img.franco.wiki/i/85820240f7cf035236aae3e889690c46f8f0f318.svg)
On a, avec la forme généralisée (FG2) de l'équation d'état cubique :

avec  la dérivée partielle du terme de cohésion par rapport à la température à volume et composition constants, or le paramètre ne dépend pas du volume : . On suppose ici que seul le paramètre dépend de la température.
la dérivée partielle du terme de cohésion par rapport à la température à volume et composition constants, or le paramètre ne dépend pas du volume : . On suppose ici que seul le paramètre dépend de la température.
En conséquence :
![{\displaystyle n{\bar {S}}^{\text{RES}}=\int _{+\infty }^{V}\left[{nR \over V-nb}-{n^{2}a_{T}^{\prime } \over \left(V-nbr_{1}\right)\left(V-nbr_{2}\right)}-{nRT \over V}\right]\,\mathrm {d} V+nR\ln Z}](https://img.franco.wiki/i/aa5c6cea28c2633aa8fbc0a6f7d5b0b599815e19.svg)
![{\displaystyle =nR\int _{+\infty }^{V}\left[{1 \over V-nb}-{1 \over V}\right]\,\mathrm {d} V-n^{2}a_{T}^{\prime }\int _{+\infty }^{V}{1 \over \left(V-nbr_{1}\right)\left(V-nbr_{2}\right)}\,\mathrm {d} V+nR\ln Z}](https://img.franco.wiki/i/9a79dd66a4ab344acadc22ffaf36ac2863eb965e.svg)

Avec par définition :
en réarrangeant on trouve :

- Calcul de l'énergie interne résiduelle molaire
Pour l'énergie interne résiduelle molaire, il suffit de considérer la définition :

- Calcul du volume résiduel molaire
Pour le volume résiduel molaire, il suffit de considérer la définition :

avec  le volume molaire du gaz parfait aux mêmes pression, température et composition que le mélange réel.
le volume molaire du gaz parfait aux mêmes pression, température et composition que le mélange réel.
- Calcul de l'enthalpie résiduelle molaire
Pour l'enthalpie résiduelle molaire, il suffit de considérer la définition :

- Calcul de l'enthalpie libre résiduelle molaire
Pour l'enthalpie libre résiduelle molaire, il suffit de considérer la définition :






























![{\displaystyle {\bar {V}}-b={RT \over P}\left[1-{\left({\bar {V}}-b\right){a \over RT} \over \left({\bar {V}}-br_{1}\right)\left({\bar {V}}-br_{2}\right)}\right]}](https://img.franco.wiki/i/d26b27366ab21b23de4b1b1a88515552771f21f8.svg)
![{\displaystyle \lim _{P\to +\infty }\left[{\bar {V}}-b\right]=0}](https://img.franco.wiki/i/23fd93dbbc652dda0d39035bd37084d567625350.svg)



