Super-famille de protéines
Une superfamille (ou super-famille) de protéines est le regroupement le plus large (clade) de protéines pour lesquelles il est possible d'identifier un ancêtre commun par homologie. Cet ancêtre commun est généralement déduit par alignement structurel (en)[1] et similitude mécanique, même lorsque aucune similitude entre les séquences n'est détectable[2]. Les super-familles contiennent généralement plusieurs familles de protéines présentant des similitudes de séquences au sein de ces familles. On utilise souvent le terme de clan pour les super-familles de peptidases en fonction de leur classification MEROPS[2].
Identification

En bas : conservation des séquences pour le même alignement, les flèches indiquant les résidus des triades catalytiques ; alignement sur la base des structures par l'algorithme DALI (FSSP).
Plusieurs méthodes permettent d'identifier les super-familles de protéines.
Homologie séquentielle

Historiquement, l'observation de similitudes entre plusieurs séquences d'acides aminés a été la méthode la plus courantes pour déduire l'existence d'une homologie entre protéines[3]. La similitude entre séquences est considérée être un bon indicateur de parenté entre protéines car de telles similitudes ont davantage de chances d'être le résultat d'une duplication de gènes suivie d'une évolution divergente plutôt qu'être le résultat d'une évolution convergente. La séquence des acides aminés est généralement mieux conservé que la séquence de l'ADN des gènes en raison de la dégénérescence du code génétique, ce qui en fait une méthode de détection plus sensible. La substitution d'un acide aminé par un autre présentant des propriétés semblables (taille, charge électrique, caractère hydrophile ou hydrophobe notamment) est généralement sans effet sur la fonction d'une protéine, de sorte que les séquences les mieux conservées correspondent aux régions qui sont fonctionnellement déterminantes pour une protéine, comme les sites de liaison et le site actif d'une enzyme, car ces régions sont moins tolérantes aux modifications de séquences.
Le recours à la similitude séquentielle pour déduire l'homologie évolutive présente cependant plusieurs limitations. Il n'existe pas de niveau minimum de similitude entre séquences garantissant que les structures produites seront identiques. Au terme de longues périodes d'évolution, des protéines apparentées peuvent ne plus présenter de similitude séquentielle détectable entre elles. Des séquences présentant de nombreuses insertions et délétions peuvent également être difficiles à aligner, ce qui complique l'identification des régions homologues correspondantes. Ainsi, dans le clan PA (en) des peptidases, aucun des résidus n'est conservé à travers la super-famille, même pas ceux de la triade catalytique. A contrario, les familles qui constituent les super-familles sont définies sur la base de leur alignement de séquences, comme c'est le cas pour la famille C04 du clan PA.
Néanmoins, la similitude séquentielle demeure la méthode la plus employée pour déterminer les relations entre protéines car le nombre de séquences connues est très largement supérieur au nombre de structures tertiaires connues[4]. Elle permet notamment de définir les contours des super-familles en l'absence d'informations structurelles.
Similitude structurelle
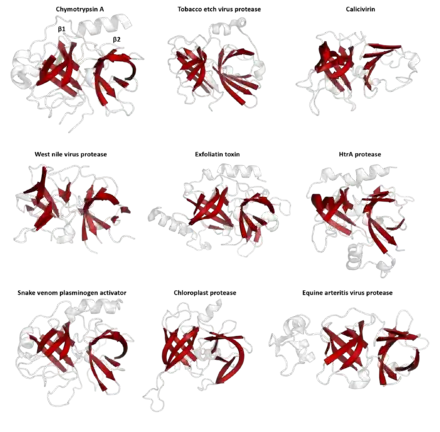
Les structures sont bien plus conservées que les séquences au cours de l'évolution, de sorte que des protéines ayant des structures extrêmement semblables peuvent avoir des séquences totalement différentes. Après de longues périodes d'évolution, très peu de résidus sont conservés, alors que les éléments de structure secondaire et les motifs de structure tertiaire demeurent hautement conservés. Les modifications conformationnelles peuvent également être conservées, comme on l'observe dans la super-famille des serpines. Par conséquent, la structure tertiaire peut permettre de détecter une homologie entre protéines même lorsque plus aucune homologie structurelle ne subsiste entre elles.
Des algorithmes d'alignement structurel (en) tels que DALI comparent la structure tridimensionnelle des protéines avec une protéine particulière afin d'identifier les repliements semblables.
Il arrive cependant dans de rares cas que des protéines évoluent de telle sorte qu'elles ne présentent plus ni similitude séquentielle ni similitude structurelle, et leur homologie ne peut alors être déduite qu'à l'aide d'autres méthodes.
Similitude mécanique
Le mécanisme réactionnel de catalyse enzymatique est généralement conservé au sein d'une super-famille bien que la spécificité vis-à-vis des substrats puisse considérablement varier. Les résidus catalytiques tendent à se présenter dans le même ordre dans la séquence des protéines. Ainsi, les peptidases du clan PA (en) utilisent toutes le même mécanisme catalytique malgré leur évolution divergente qui a modifié jusqu'aux résidus de leur triade catalytique. Cependant, l'existence d'une similitude mécanique ne suffit pas à déduire une relation de parenté entre protéines dans la mesure où certains mécanismes catalytiques peuvent résulter d'une évolution convergente intervenue de manière indépendante à des époques différentes ; de telles enzymes sont dans ce cas rangées dans des super-familles distinctes[5].
Application aux recherches sur l'évolution
Les super-familles de protéines représentent les limites actuelles de notre capacité à remonter aux ancêtres communs[6]. Ce sont les regroupements de protéines les plus larges qu'il est actuellement possible d'établir sur la base d'éléments de similitude directs. Elles permettent par conséquent de remonter aux étapes d'évolution les plus anciennes qu'il soit possible d'étudier. Certaines d'entre elles sont présentes parmi tous les règnes du vivant, indiquant qu'elles remontent au moins au dernier ancêtre commun universel[7] (LUCA).
Les protéines d'une même super-famille peuvent se retrouver chez différentes espèces, la forme ancestrale de ces protéines étant dans ce cas celle de l'espèce ancestrale commune à toutes ces espèces : il s'agit dans ce cas d'une relation d'orthologie ; a contrario, elles peuvent se trouver au sein d'une même espèce et provenir d'une protéine ancestrale unique dont le gène a évolué par duplication au sein du génome : il s'agit dans ce cas d'une relation de paralogie.
Diversification
La majorité des protéines contiennent plusieurs domaines. Environ 66 % à 80 % des protéines d'eucaryotes ont plusieurs domaines tandis qu'environ 40 % à 60 % des protéines de procaryotes ont plusieurs domaines[3]. De nombreuses super-familles de domaines protéiques ont fini par se mélanger avec le temps, de sorte qu'il est très rare de trouver des super-familles toujours isolées. Cependant, le nombre de combinaisons de domaines observé dans la nature demeure faible par rapport au nombre de combinaisons possibles, ce qui laisse penser que la sélection agit sur toutes les combinaisons[3].
Notes et références
- (en) Liisa Holm et Päivi Rosenström, « Dali server: conservation mapping in 3D », Nucleic Acids Research, vol. 38, no Supplement 2, , W545-W549 (PMID 20457744, PMCID 2896194, DOI 10.1093/nar/gkq366, lire en ligne)
- (en) Neil D. Rawlings, Alan J. Barrett et Alex Bateman, « MEROPS: the database of proteolytic enzymes, their substrates and inhibitors », Nucleic Acids Research, vol. 40, no D1, , D343-D350 (PMID 22086950, PMCID 3245014, DOI 10.1093/nar/gkr987, lire en ligne)
- (en) Jung-Hoon Han, Sarah Batey, Adrian A. Nickson, Sarah A. Teichmann et Jane Clarke, « The folding and evolution of multidomain proteins », Nature Reviews Molecular Cell Biology, vol. 8, no 4, , p. 319-330 (PMID 17356578, DOI 10.1038/nrm2144, lire en ligne)
- (en) Shashi B. Pandit, Dilip Gosar, S. Abhiman, S. Sujatha, Sayali S. Dixit, Natasha S. Mhatre, R. Sowdhamini et N. Srinivasan, « SUPFAM—a database of potential protein superfamily relationships derived by comparing sequence-based and structure-based families: implications for structural genomics and function annotation in genomes », Nucleic Acids Research, vol. 30, no 1, , p. 289-293 (PMID 11752317, PMCID 99061, DOI 10.1093/nar/30.1.289, lire en ligne)
- (en) Andrew R. Buller et Craig A. Townsend, « Intrinsic evolutionary constraints on protease structure, enzyme acylation, and the identity of the catalytic triad », Proceedings of the National Academy of Sciences of the United States of America, vol. 110, no 8, , E653-E661 (PMID 23382230, PMCID 3581919, DOI 10.1073/pnas.1221050110, lire en ligne)
- (en) Boris E. Shakhnovich, Eric Deeds, Charles Delisi et Eugene Shakhnovich, « Protein structure and evolutionary history determine sequence space topology », Genome Research, vol. 15, no 3, , p. 385-392 (PMID 15741509, PMCID 551565, DOI 10.1101/gr.3133605, lire en ligne)
- (en) Juan A. G. Ranea, Antonio Sillero, Janet M. Thornton et Christine A. Orengo, « Protein Superfamily Evolution and the Last Universal Common Ancestor (LUCA) », Journal of Molecular Evolution, vol. 63, no 4, , p. 513-525 (PMID 17021929, DOI 10.1007/s00239-005-0289-7, lire en ligne)