Lymphohistiocytose hémophagocytaire
Cet article traite de la forme familiale de la lymphohistiocytose hémophagocytaire, maladie génétique à ne pas confondre avec la lymphohistiocytose hémophagocytaire secondaire.
| Spécialité | Hématologie |
|---|
| CISP-2 | B99 |
|---|---|
| CIM-10 | D76.1 |
| CIM-9 | 288.4 |
| OMIM | 267700 603553 608898 603552 |
| DiseasesDB | 31418 |
| eMedicine | 986458 |
| MeSH | D051359 |
![]() Mise en garde médicale
Mise en garde médicale
La lymphohistiocytose hémophagocytaire ou syndrome d'activation macrophagique est une maladie rare du groupe des histiocytoses] non langerhansiennes et non malignes. Ce syndrome est marqué par une dangereuse prolifération de macrophages bénins activés, avec une hémophagocytose intense, et un défaut de l’activité cytotoxique des lymphocytes NK et des lymphocytes T CD8+, et hypercytokinémie (tempête de cytokines). La forme familiale ou LHF (lymphohistiocytose familiale), héréditaire et à transmission autosomique récessive, rare est rapidement fatale. Elle touche 1 naissance sur 50000. Elle apparaît généralement dès les premiers mois de la vie et le diagnostic peut être difficile ;
Historique
En 1939, la réticulose médullaire histiocytaire a été mise en évidence, associant une altération de l’état général de l’organisme, à une organomégalie et une pancytopénie périphérique[1].
Les premiers cas de lymphohistiocytose hémophagocytaire familiale (LHF) ont été décrits par Farquhar et Claireaux en 1952. En 1966, Rappaport décrit une prolifération systémique d’histiocytes atypiques et regroupe ses observations sous le terme d’histiocytose maligne. En 1979, Risdall rapporte que le syndrome hémophagocytaire peut être aussi associé à une infection virale.
Épidémiologie
Cette maladie est rare. L'essentiel des cas est pédiatrique (environ un enfant sur un million est touché[2]) ; chez l'adulte, la moitié des cas sont secondaires à un cancer, essentiellement un lymphome non hodgkinien[3], avec un pronostic grave[4].
Tableau clinique
Les signes biologiques et cliniques sont très divers.
Les symptômes généraux évoluent dès leur apparition. généralement une forte fièvre s’installe, avec sueurs et frissons.
L'examen clinique monte généralement une splénomégalie (grosse rate) ou une hépatomégalie (gros foie). Celles-ci sont dues à une infiltration par le contingent histiocytaire. Chez l'enfant, la dilatation de ces organes peut prendre un aspect pseudo-tumoral.
De possibles atteintes neurologiques sont l’ataxie, des crises convulsives, une irritabilité, une encéphalopathie.
Une atteinte cutanée est fréquente (purpura, œdème, ictère le plus souvent, et parfois éruptions cutanées de type érythème).
Le poumon est touché dans 20 à 30 % des cas (infiltrats pulmonaires visibles sur la radiographie du thorax.
De possibles signes digestifs sont des hémorragies digestives (causée par une probable thrombopénie) et des diarrhées.
Une atteinte rénale ou cardiaque peuvent survenir.
In fine une défaillance multi-organe entraînait autrefois la mort des patients. Malgré le progrès des traitements, une partie des patients en meurent encore.
Diagnostic
Il se fait sur la base de la présence de 5 au moins des symptômes suivants[5] :
- Fièvre
- Splénomégalie
- Absence de malignité
- Cytopénies touchant au moins deux lignées hématopoïétiques :
- hémoglobine < 9 g/dL (enfants de moins de 4 semaines < 10 g/dL)
- plaquettes < 100 x 109 /L
- neutrophiles < 1,0 x 109 /L
- Triglycérides ≥ 265 mg/dL
- Fibrinogène < 1,5 g/L
- Niveau élevé des lactates désydrogènases chez l'adulte[6]
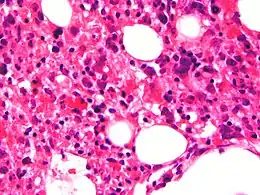
- Hémophagocytose dans la moelle osseuse ou dans la rate voire dans les ganglions lymphatiques. Ces cellules sont des macrophages avec présence d'éléments hématopoïétique dans le cytoplasme. Elles sont inconstamment retrouvées[7] et peuvent être également présentes en dehors de la lymphohistiocytose hémophagocytaire[8].
- Taux bas ou absence de l’activité des NK
- Ferritine ≥ 500 µg/L et pouvant dépasser 10 000 µg/L, ce qui est très évocateur du diagnostic chez l'enfant[9] mais moins chez l'adulte[10].
- CD25 soluble ≥ 2400 U/mL
Dans les formes familiales de LH, on recherche bien sûr les mutations sur les gènes connus pour être associés à la maladie (diagnostic moléculaire).
Mécanismes moléculaires et physiopathologiques
La lymphohistiocytose est caractérisée par un défaut de l’activité cytotoxique des lymphocytes NK et lymphocytes TCD8+, une suractivation des macrophages et une hypercytokinémie.
Les cas familiaux de lymphohistiocytoses hémophagocytaires proviennent de mutations de gènes codant des molécules essentielles au processus d’exocytose des granules lytiques et donc à la cytotoxicité :
- gène PRF1 : gène de la perforine (protéine cytolytique sécrétée par les lymphocytes T CD8+) localisée sur le chromosome 10q21-22 est muté chez 15 à 50 % des patients présentant une LHF.
- gène UNC13D : gène qui code la protéine Munc13-4 localisé sur le chromosome 17q25 normalement responsable de l’arrimage des granules cytolytiques en préparation pour la fusion avec la membrane plasmique. Environ 15 à 30 % des patients ayant une LHF présente cette mutation.
- gène STX11 : gène localisé sur le chromosome 6q24 codant la syntaxine 11 qui participe à la fusion des membranes vésiculaire et cellulaire. Cette mutation est présente chez environ 20 % des patients d’origine turque présentant une LHF.
Le résultat de ces déficiences est double : mauvaise élimination des antigènes, donc hyperstimulation du système immunitaire avec une forte sécrétion de cytokines ; et défaut de régulation des macrophages par les cellules NK. Il y a donc accumulation de lymphohistiocytes dans les organes et notamment dans les espaces périvasculaires et les méninges du cerveau. La tempête de cytokine cause la plupart des signes cliniques et biologiques observés de la LHF ; la variation d’interleukines en particulier de l'interleukine 1 cause la fièvre. Un niveau élevé de TNF-α et d’interférons-γ (INF-γ) entraînent une intense fatigue, une sudation importante ainsi qu’une pancytopénie.
Des indices laissent penser que, même hors des formes familiales, des facteurs génétiques sous-jacents pourraient, une fois activés par certaines maladies et/ou des déclencheurs environnementaux (infection, cancer, trouble auto-immun et/ou auto-inflammatoire) induire une sHLH[11]. Les progrès des tests génétiques attendus dans les années 2020 devraient permettre de tester un plus grand nombre de gènes potentiellement associés à la HLH primaire (pHLH), dont en séquençant l'exome et le génome dans leur totalité. Si cette hypothèse est confirmée, la HLH serait encore plus complexe qu'on le pensait, son traitement et sa définition pourrait évoluer, ainsi que ses traitements, selon qu'il s'agisse de complication grave d'infections ou de formes familiales (nécessitant des transplantations)[11].
Traitement
Il est déterminé selon la sévérité des symptômes et de la cause de la maladie, de manière à contrôler les symptômes.
- Pour les formes familiales (aussi dites primaires) : un traitement immunosuppresseur précède une greffe de moelle osseuse. L’étoposide, en association avec des corticoïdes, a été utilisé avec succès[12].
- Pour les formes secondaires, il faut aussi et surtout traiter le facteur déclenchant (infections notamment) tout en administrant un traitement immunosuppresseur.
Les médicaments utilisées sont :
- Corticostéroïde
- Ciclosporine A
- Etoposide
- Injection d’immunoglobulines en intraveineuse.
Une allogreffe de mœlle osseuse peut être nécessaire.
Voir aussi
Articles connexes
Notes et références
- Scott RB, Robb-Smith AH, Histiocytic medullary reticulocytosis, Lancet, 1939;2:194-198
- Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X, Adult haemophagocytic syndrome, Lancet, 2014;383:1503-1516
- Rivière S, Galicier L, Coppo P et al. Reactive hemophagocytic syndrome in adults: a retrospective analysis of 162 patients, Am J Med, 2014;127:1118-1125
- Parikh SA, Kapoor P, Letendre L, Kumar S, Wolanskyj AP, Prognostic factors and outcomes of adults with hemophagocytic lymphohistiocytosis, Mayo Clin Proc, 2014;89:484-492
- Henter JI, Horne A, Aricó M et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis, Pediatr Blood Cancer, 2007;48:124-131
- Hejblum G, Lambotte O, Galicier L et al. A Web-based Delphi study for eliciting helpful criteria in the positive diagnosis of hemophagocytic syndrome in adult patients, PLoS One, 2014;9:e94024-e94024
- Gupta A, Weitzman S, Abdelhaleem M, The role of hemophagocytosis in bone marrow aspirates in the diagnosis of hemophagocytic lymphohistiocytosis, Pediatr Blood Cancer, 2008;50:192-194
- Suster S, Hilsenbeck S, Rywlin AM, Reactive histiocytic hyperplasia with hemophagocytosis in hematopoietic organs: a reevaluation of the benign hemophagocytic proliferations, Hum Pathol, 1988;19:705-712
- Allen CE, Yu X, Kozinetz CA, McClain KL, Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis, Pediatr Blood Cancer, 2008;50:1227-1235
- Schram AM, Campigotto F, Mullally A et al. Marked hyperferritinemia does not predict for HLH in the adult population, Blood, 2015;125:1548-1552
- (en) Grant S. Schulert et Kejian Zhang, « Genetics of Acquired Cytokine Storm Syndromes », dans Cytokine Storm Syndrome, Springer International Publishing, (ISBN 978-3-030-22094-5, DOI 10.1007/978-3-030-22094-5_7, lire en ligne), p. 113–129
- Bergsten E, Horne A, Aricó , et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study, Blood, 2017;130:2728-2738