Fluoration (chimie)
La fluoration est une réaction chimique d'halogénation qui ajoute un ou plusieurs atomes de fluor sur une molécule. Cette réaction peut être une addition pour les alcènes ou une substitution pour les alcanes ou les composés aromatiques.
Plusieurs voies de synthèse existent pour la fluoration :
- la substitution radicalaire ;
- la substitution nucléophile ;
- l'addition électrophile ;
- la fluoration électrochimique ;
- la diazotation.
Substitution radicalaire
La substitution radicalaire est composée de trois étapes : amorçage, propagation et terminaison. Le difluor forme facilement des radicaux : l'énergie de dissociation homolytique est de 37 kJ/mol[1]. Ceci permet un amorçage facile de radicaux via activation UV ou par la chaleur. L'étape de propagation est composée de deux parties :
- réaction entre un radical fluoré et un hydrogène de la molécule à fluorer ;
- réaction entre le radical aliphatique et une molécule de difluor.
Ces deux réactions sont fortement exothermiques dans le cas de la fluoration (respectivement −30 kJ/mol et −73 kJ/mol[1]).
- 1)
- 2)


Cette forte exothermicité rend la réaction explosive et difficile à contrôler. Pour réduire les risques, le difluor est dilué dans des gaz inertes comme le diazote ou le gaz carbonique, le substrat organique dilué dans un solvant ou la température de réaction abaissée[2]. De plus, cette forte réactivité se traduit par une faible sélectivité de la fluoration.
Ce procédé est utilisé à l'échelle industrielle pour produire le fluorouracile[3].
Substitution nucléophile
Avec oxydation
En utilisant des fluorures métalliques, tels le trifluorure de cobalt ou le difluorure d'argent, le transfert d'un fluor s'effectue de manière moins réactive. Le complexe métallique peut être régénéré après réaction avec du difluor[2].
- 1)
- 2)


Ce procédé est utilisé tant pour fluorer des composés aliphatiques qu'aromatiques[3].
Sans oxydation
Ce procédé consiste à substituer un chlore déjà présent sur la molécule par un fluor à l'aide de fluorure d'hydrogène. La réaction peut avoir lieu en phase liquide ou gazeuse avec ou sans catalyseur[2]. Pour la phase liquide, les catalyseurs les plus fréquents sont le pentachlorure d'antimoine[3] alors qu'en phase gazeuse, les fluorures métalliques tels le fluorure d'aluminium sont utilisés.
Ce procédé est utilisé pour fluorer les composés aromatiques (et dans ce cas est parfois connu sous le nom de fluoration Halex) qui doivent être toutefois activés à l'aide d'autres groupes fonctionnels et avec comme source de fluor des fluorures alcalins tels que le fluorure de potassium ou le fluorure de sodium[3]. Le fluorure de césium est le plus réactif, mais dangereux à l'échelle industrielle et le fluorure de rubidium trop cher et peu disponible[3].
Addition électrophile
L'addition de fluor sur des alcènes et des alcynes se fait à l'aide de fluorure d'hydrogène. La réaction a lieu à basses températures sauf pour l'éthylène ou l'acétylène qui a lieu à 90 °C[2].
Fluoration électrochimique
Deux procédés électrochimiques ont été développés pour la fluoration de composés organiques : le procédé Simons et le procédé CAVE-Phillips.
Procédé Simons
Le procédé Simons a été inventé par le chimiste américain Joseph Simons et breveté en 1946 par l'entreprise 3M[3]. Les réactifs organiques sont dissous dans le fluorure d'hydrogène anhydre. La réaction de fluoration a lieu à l'anode en nickel de la cellule électrochimique. En principe, tous les hydrogènes sont substituables par le fluor et toutes les liaisons multiples carbone-carbone peuvent être saturées. Des coproduits tel l'hydrogène formé à la cathode sont évacués sous forme gazeuse et condensés. Les produits fluorés sont en général insolubles dans le solvant et sont récupérés au fond de la cellule[3]. Ce procédé est utilisé principalement pour la synthèse de composés complètement fluorés[3].
Procédé CAVE-Phillips
Ce procédé fut inventé et développé par l'entreprise Phillips Petroleum et fut désigné par l'entreprise 3M sous le nom de CAVE (Carbon Anode Vapor phase Electrochemical fluorination, fluoration électrochimique en phase gazeuse par anode de carbone)[3].
L'électrolyte est constitué d'un mélange de bifluorure de potassium et de fluorure d'hydrogène qui forme un composé KF•2 HF. L'ensemble de la cellule en acier est utilisé comme cathode alors que l'anode est formée de carbone amorphe non-graphitique. La cellule est alimentée en fluorure d'hydrogène et en réactif sous forme gazeuse ou facilement volatil et les produits ainsi que l'hydrogène sont évacués sous forme de gaz. Les produits sont un mélange de composés fluorés à divers degrés[3].
L'électrolyte ne mouillant pas l'anode, c'est par la formation de fluor élémentaire et par un mécanisme radicalaire que la réaction a lieu[3].
Diazotation
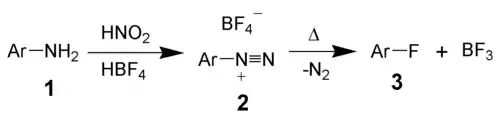
Réaction de Balz-Schiemann
La réaction de Balz-Schiemann a été la première méthode utilisée pour l'introduction d'un fluor sur un cycle aromatique. Toutefois, elle nécessite un excellent contrôle du procédé et n'a été que tardivement utilisée à l'échelle industrielle[3].
Cette synthèse se déroule en deux étapes :
- formation d'un sel tétrafluoroborate d'arènediazonium à partir d'une amine aromatique ;
- décomposition du sel pour donner le fluorure aromatique.
HF diazotation - dédiazoniation
La réaction de Balz-Schiemann a été testée avec plusieurs contre-ions en lieu et place du tétrafluoroborate : fluorure, hexafluorophosphate, hexafluoroantimonate, hexafluorosilicate ou hexafluoroarsonate. Seul le fluorure a conduit à une application industrielle[3].
La diazotation se fait à l'aide de fluorure de nitrosyle généré in situ par réaction entre le fluorure d'hydrogène et le nitrite de sodium. Le fluorure de nitrosyle réagit avec l'amine aromatique pour former le sel fluorure d'arènediazonium. La décomposition finale donne le fluorobenzène avec dégagement de diazote[3].
1)

2)

3)

L'avantage de cette voie de synthèse sur la réaction de Balz-Schiemann est qu'il n'y a pas de besoin d'isoler le sel intermédiaire et que la décomposition ne produit pas de trifluorure de bore qu'il faut récupérer[3].
Notes et références
- (en) K. Peter C. Vollhardt et Neil E. Shore, Organic chemistry, New York, W. H. Freeman, , 2e éd., 1156 p. (ISBN 978-0-7167-2010-2), p. 85
- (en) Günter Siegemund, Werner Schwertfeger, Andrew Feiring, Bruce Smart, Fred Behr, Herward Vogel, Blaine McKusick, Fluorine Compounds, Organic, Wiley-VCH Verlag, coll. « Ullmann's Encyclopedia of Industrial Chemistry »,
- (en) R. E. Banks, B. E. Smart et J. C. Tatlow, Organofluorine Chemistry : Principles and Commercial Applications, New York, Plenum Press, , 644 p. (ISBN 0-306-44610-3)