Cellule bêta
Les cellules bêta (β-cells pour les anglophones) sont l'un des types cellulaires du pancréas, et plus particulièrement des îlots de Langerhans. Chez l'être humain, elles représentent 50 à 70 % des cellules de ces îlots[1]. Ce sont elles qui synthétisent et sécrètent l'insuline et l'amyline. Chez les patients atteints de diabète de type 1, la masse et la fonction de ces cellules diminue, entraînant une sécrétion insuffisante d'insuline et une hyperglycémie[2].
Rôle
Les cellules bêta produisent et libèrent dans le sang de manière endocrine trois hormones essentielles qui réduisent la glycémie par différents mécanismes :
- L'insuline, l'hormone hypoglycémiante qui régule la glycémie (le taux de glucose sanguin). Les cellules bêta la produisent en continu et en stockent aussi, répondant à l'augmentation de la glycémie par sa libération rapide dans le sang ;
- Le peptide C (composé dérivé de l'insuline). Ce peptide est une hormone qui semble jouer un rôle contre la neuropathie et d'autres symptômes liés à la détérioration vasculaire associée au diabète sucré[3].
Il est sécrété et injecté dans la circulation sanguine en quantités équimolaires à l'insuline.
La mesure du niveaux de peptide C est un moyen d'estimer la masse de cellules bêta fonctionnelles[4] ; - L'amyline, ou IAPP (acronyme d' islet amyloid polypeptide)[5], inhibiteur de l'augmentation de la charge du sang en nutriments. Ses effets sont donc synergiques de ceux de l'insuline.
Les cellules bêta réagissent rapidement aux pics de glucose dans le sang, en sécrétant une partie de leur insuline et de l'amyline stockées tout en en produisant plus[6].
Synthèse de l'insuline
Tout excès de glucose dans le sang stimule l'introduction d'insuline dans le sang, et augmente simultanément la biosynthèse de la proinsuline (en), principalement par le contrôle de la traduction[6]. Dans l'organisme des mammifères, les cellules bêta sont l'unique lieu de synthèse de l'insuline [4] ;
Le gène de l'insuline est d'abord transcrit en ARNm et traduit en préproinsuline[6]. Après traduction, le précurseur de la préproinsuline contient un peptide signal N-terminal qui permet la translocation dans le réticulum endoplasmique rugueux (RER)[7].
Dans le RER, le peptide signal est clivé pour former de la proinsuline[7]. Ensuite, le repliement de la proinsuline se produit en formant trois liaisons disulfure[7]. Après le repliement des protéines, la proinsuline est transportée vers l'appareil de Golgi et pénètre dans les granules d'insuline immatures où la proinsuline est clivée pour former l'insuline et le peptide C[7]. Après maturation, ces vésicules de sécrétion contiennent de l'insuline, du peptide C et de l'amyline jusqu'à ce que le calcium déclenche l'exocytose du contenu des granules[6].
Grâce au traitement traductionnel, l'insuline est codée en tant que précurseur de 110 acides aminés, mais est sécrétée en tant que protéine de 51 acides aminés[7].
Sécrétion d'insuline
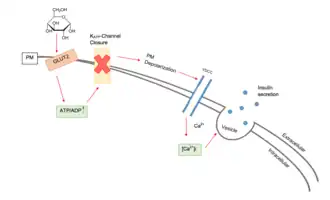
Dans les cellules bêta, la libération d'insuline est principalement stimulée par le glucose présent dans le sang[6] ; plus le taux de glucose circulant augmente (après l'ingestion d'un repas par exemple), l'insuline est sécrétée de manière dose-dépendante[6] - [8]. Le « modèle de consensus » du GSIS comprend quatre éléments clés : l'absorption glucose dépendante de GLUT2, le métabolisme du glucose, la fermeture des canaux KATP et l'ouverture des canaux calciques voltage-dépendants provoquant la fusion des granules d'insuline et l'exocytose[9].
Les canaux calciques voltage-dépendants et les canaux ioniques potassiques sensibles à l'adénosine triphosphate (ATP) sont intégrés dans la membrane plasmique des cellules bêta[9] - [10]. Ces canaux ioniques potassiques sensibles à l'ATP sont normalement ouverts et les canaux ioniques calcium sont normalement fermés[6]. Les ions potassium diffusent hors de la cellule, vers le bas de leur gradient de concentration, rendant l'intérieur de la cellule plus négatif par rapport à l'extérieur (car les ions potassium portent une charge positive)[6]. Au repos, cela crée une différence de potentiel à travers la membrane de surface cellulaire de -70 mV [11].
Quand le taux de glucose est élevée hors de la cellule, des molécules de glucose pénètrent dans la cellule par « diffusion facilitée », en diminuant le gradient de concentration via le transporteur GLUT2[12].
Comme les cellules bêta utilisent la glucokinase pour catalyser la première étape de la glycolyse, le métabolisme ne se produit qu'autour de la glycémie physiologique et au-dessus[6]. Le métabolisme du glucose produit de l'ATP, ce qui augmente le rapport ATP/ADP [13].
Les canaux ioniques potassiques sensibles à l'ATP se ferment lorsque ce rapport augmente<refq name=Ashcroft1990/>. Ceci signifie que les cations potassium ne peuvent plus diffuser hors de la cellule[14]. En conséquence, la différence de potentiel à travers la membrane devient plus positive (à mesure que les ions potassium s'accumulent à l'intérieur de la cellule)[10]. Ce changement de différence de potentiel ouvre les canaux calciques voltage-dépendants, ce qui permet aux ions calcium de l'extérieur de la cellule de se diffuser dans leur gradient de concentration[10]. Lorsque les ions calcium pénètrent dans la cellule, ils provoquent le déplacement et la fusion de vésicules contenant de l'insuline vers la membrane de surface cellulaire, libérant de l'insuline par exocytose dans la veine porte hépatique[15] - [16].
Pathologies
Diabète de type 1
Une forme de diabète, le diabète de type I aussi dit insulinodépendant, est causé par une destruction auto-immune des cellules bêta productrices d'insuline[17].
Le processus de destruction des cellules bêta commence par l'activation des cellules présentatrices d'antigène (CPA) par l'insulite. Les APC déclenchent alors l'activation des cellules T auxiliaires CD4+ et la libération de chimiokines/cytokines. Les cytokines activent alors les lymphocytes cytotoxiques T CD8+, ce qui entraîne la destruction des cellules bêta[17]. La destruction de ces cellules empêche le corps de bien répondre aux niveaux de glucose dans le corps, ce qui rend presque impossible le bon ajustement des taux de glucose et de glucagon sanguins[18]. Le corps autodétruit 70 à 80 % des cellules bêta, ne laissant que 20 à 30 % des cellules fonctionnelles[19].
Cela peut entraîner une hyperglycémie chez le patient, laquelle entraîne d'autres affections indésirables à court et à long terme[20]. Ce type de diabète peut initialement êtree contrôlé par des prises régulières d'insuline et le maintien d'un régime alimentaire approprié[20]. Cependant, ces méthodes peuvent être fastidieuses et lourdes à exécuter en continu au quotidien[20].
Diabète de type 2
Le diabète sucré de type 2 (aussi dénommé diabète non insulino-dépendant ou diabète d'hyperglycémie chronique, est principalement causé par la génétique et le développement du syndrome métabolique[2]. Dans ce cas, les cellules bêta peuvent encore sécréter de l'insuline, mais c'est le corps qui développe une résistance à l'insuline, il y répond de moins en moins bien. On pense que cela est dû au déclin de récepteurs spécifiques à la surface du foie, des cellules adipeuses et musculaires qui perdent leur capacité à répondre à l'insuline qui circule dans le sang[21] - [22]. Dans un effort pour sécréter suffisamment d'insuline pour surmonter la résistance croissante à l'insuline, les cellules bêta augmentent leur fonction, leur taille et leur nombre[6]. L'augmentation de la sécrétion d'insuline entraîne une hyperinsulinémie, mais les taux de glycémie restent dans leur plage normale en raison de la diminution de l'efficacité de la signalisation de l'insuline[6]. Cependant, les cellules bêta s'épuisent à force d'être surstimulées, ce qui entraîne une réduction de 50 % de la fonction ainsi qu'une diminution de 40 % du volume des cellules bêta[17]. À ce stade, il n'est pas possible de produire et de sécréter suffisamment d'insuline pour maintenir les niveaux de glucose dans le sang dans leur fourchette normale, ce qui provoque un diabète de type 2 manifeste[17].
Tumeur
Le Nésidioblastome (ou insulinome est une tumeur rare issue de cellules bêta.
Les insulinomes sont généralement bénins, mais peuvent avoir des conséquences hormonales et médicales graves, voire mortelles en raison d'attaques récurrentes et prolongées d'hypoglycémie[23].
Médicaments ciblant la cellule bêta
Beaucoup des médicaments traitant le diabète visent à modifier la fonction de la cellule bêta.
Les sulfonylurées sont des sécrétagogues de l'insuline (en fermant les canaux potassiques sensibles à l'ATP, ils provoquant ainsi la libération d'insuline)[24] - [25]. Ces médicaments sont connus pour provoquer une hypoglycémie et peuvent entraîner une défaillance des cellules bêta en raison d'une surstimulation[2]. Les versions de deuxième génération de sulfonylurées ont une action plus courte et sont moins susceptibles de provoquer une hypoglycémie[25]. Les agonistes des récepteurs du GLP-1 stimulent la sécrétion d'insuline en simulant l'activation du système incrétine endogène du corps[25]. Le système incrétine agit comme une voie d'amplification de la sécrétion d'insuline[25]. Les inhibiteurs de la DPP-4 bloquent l'activité de la DPP-4, ce qui augmente la concentration postprandiale des hormones gastro-intestinales incrétines (GLP-1 pour glucagon-like peptide-1 et GIP pour Peptide insulinotrope dépendant du glucose ou glucose-dependent insulinotropic peptide), ce qui a pour effet d'augmente la sécrétion d'insuline[25].
Recherche
Techniques expérimentales
De nombreux chercheurs étudient la pathogenèse du diabète et de la défaillance des cellules bêta. Les outils utilisés pour étudier la fonction des cellules bêta se développent rapidement avec la technologie.
Ainsi, la transcriptomique a permis aux chercheurs de mieux analyser la transcription des gènes dans les cellules bêta pour rechercher des gènes liés au diabète[2]. Un mécanisme plus courant d'analyse de la fonction cellulaire est l'imagerie calcique. Les colorants fluorescents se lient au calcium et permettent une imagerie in vitro de l'activité du calcium qui est directement corrélée à la libération d'insuline[2] - [26]. Un dernier outil utilisé dans la recherche sur les cellules bêta sont les expériences in vivo. Le diabète sucré peut être induit expérimentalement in vivo à des fins de recherche par la streptozotocine[27] ou l'alloxane[28] qui sont spécifiquement toxiques pour les cellules bêta. Des modèles de diabète chez la souris et le rat existent également, y compris les souris ob/ob et db/db qui sont un modèle de diabète de type 2, et les souris diabétiques non obèses (NOD) qui sont un modèle pour le diabète de type 1 [29].
Notes et références
Notes
Références
- (en) Jurij Dolenšek, Marjan Slak Rupnik et Andraž Stožer, « Structural similarities and differences between the human and the mouse pancreas », Islets, vol. 7, no 1, , e1024405 (ISSN 1938-2014 et 1938-2022, PMID 26030186, PMCID PMC4589993, DOI 10.1080/19382014.2015.1024405, lire en ligne, consulté le )
- (en) Chunguang Chen, Christian M. Cohrs, Julia Stertmann et Robert Bozsak, « Human beta cell mass and function in diabetes: Recent advances in knowledge and technologies to understand disease pathogenesis », Molecular Metabolism, vol. 6, no 9, , p. 943–957 (DOI 10.1016/j.molmet.2017.06.019, lire en ligne, consulté le )
- Y. Ido, « Prevention of Vascular and Neural Dysfunction in Diabetic Rats by C-Peptide », Science, vol. 277, no 5325, , p. 563–566 (DOI 10.1126/science.277.5325.563, lire en ligne, consulté le )
- (en) Byron J. Hoogwerf et C. Goetz, « Urinary C-Peptide: A Simple Measure of Integrated Insulin Production with Emphasis on the Effects of Body Size, Diet, and Corticosteroids* », The Journal of Clinical Endocrinology & Metabolism, vol. 56, no 1, , p. 60–67 (ISSN 0021-972X et 1945-7197, DOI 10.1210/jcem-56-1-60, lire en ligne, consulté le )
- « amyline : Dictionnaire pharmaceutique - Pharmacologie et chimie des médicaments », sur www.informationhospitaliere.com (consulté le )
- (en) Brandon B. Boland, Christopher J. Rhodes et Joseph S. Grimsby, « The dynamic plasticity of insulin production in β-cells », Molecular Metabolism, vol. 6, no 9, , p. 958–973 (PMID 28951821, PMCID PMC5605729, DOI 10.1016/j.molmet.2017.04.010, lire en ligne, consulté le )
- (en) Brandon B Boland, Charles Brown, Cristina Alarcon et Damien Demozay, « β-Cell Control of Insulin Production During Starvation-Refeeding in Male Rats », Endocrinology, vol. 159, no 2, , p. 895–906 (ISSN 1945-7170, DOI 10.1210/en.2017-03120, lire en ligne, consulté le )
- (en) Mitsuhisa Komatsu, Masahiro Takei, Hiroaki Ishii et Yoshihiko Sato, « Glucose-stimulated insulin secretion: A newer perspective », Journal of Diabetes Investigation, vol. 4, no 6, , p. 511–516 (DOI 10.1111/jdi.12094, lire en ligne, consulté le )
- (en) James W. Ramadan, Stephen R. Steiner, Christina M. O’Neill et Craig S. Nunemaker, « The central role of calcium in the effects of cytokines on beta-cell function: Implications for type 1 and type 2 diabetes », Cell Calcium, vol. 50, no 6, , p. 481–490 (PMID 21944825, PMCID PMC3223281, DOI 10.1016/j.ceca.2011.08.005, lire en ligne, consulté le )
- (en) Frances M. Ashcroft et Patrik Rorsman, « ATP-sensitive K+ channels: a link between B-cell metabolism and insulin secretion », Biochemical Society Transactions, vol. 18, no 1, , p. 109–111 (ISSN 0300-5127 et 1470-8752, DOI 10.1042/bst0180109, lire en ligne, consulté le )
- (en) Patrick E MacDonald, Jamie W Joseph et Patrik Rorsman, « Glucose-sensing mechanisms in pancreatic β-cells », Philosophical Transactions of the Royal Society B: Biological Sciences, vol. 360, no 1464, , p. 2211–2225 (ISSN 0962-8436 et 1471-2970, PMID 16321791, PMCID PMC1569593, DOI 10.1098/rstb.2005.1762, lire en ligne, consulté le )
- (en) A De Vos, H Heimberg, E Quartier et P Huypens, « Human and rat beta cells differ in glucose transporter but not in glucokinase gene expression. », Journal of Clinical Investigation, vol. 96, no 5, , p. 2489–2495 (ISSN 0021-9738, DOI 10.1172/JCI118308, lire en ligne, consulté le )
- (en) Gaetano Santulli, Gennaro Pagano, Celestino Sardu et Wenjun Xie, « Calcium release channel RyR2 regulates insulin release and glucose homeostasis », Journal of Clinical Investigation, vol. 125, no 5, , p. 1968–1978 (ISSN 0021-9738, DOI 10.1172/JCI79273, lire en ligne, consulté le )
- J Keizer et G Magnus, « ATP-sensitive potassium channel and bursting in the pancreatic beta cell. A theoretical study. », Biophysical Journal, vol. 56, no 2, , p. 229–242 (ISSN 0006-3495, PMID 2673420, PMCID 1280472, lire en ligne, consulté le )
- (en) Peter Light, « The molecular mechanisms and pharmacotherapy of ATP-sensitive potassium channel gene mutations underlying neonatal diabetes », Pharmacogenomics and Personalized Medicine, , p. 145 (ISSN 1178-7066, DOI 10.2147/PGPM.S6969, lire en ligne, consulté le )
- (en) Dale S. Edgerton, Guillaume Kraft, Marta Smith et Ben Farmer, « Insulin’s direct hepatic effect explains the inhibition of glucose production caused by insulin secretion », JCI Insight, vol. 2, no 6, (ISSN 2379-3708, DOI 10.1172/jci.insight.91863, lire en ligne, consulté le )
- (en) Zhuo Fu, Elizabeth R. Gilbert et Dongmin Liu, « Regulation of Insulin Synthesis and Secretion and Pancreatic Beta-Cell Dysfunction in Diabetes », Current Diabetes Reviews, vol. 9, no 1, , p. 25–53 (DOI 10.2174/157339913804143225, lire en ligne, consulté le )
- D. L. Eizirik et T. Mandrup-Poulsen, « A choice of death - the signal-transduction of immune-mediated beta-cell apoptosis », Diabetologia, vol. 44, no 12, , p. 2115–2133 (ISSN 0012-186X et 1432-0428, DOI 10.1007/s001250100021, lire en ligne, consulté le )
- (en) A. E. Butler, R. Galasso, J. J. Meier et R. Basu, « Modestly increased beta cell apoptosis but no increased beta cell replication in recent-onset type 1 diabetic patients who died of diabetic ketoacidosis », Diabetologia, vol. 50, no 11, , p. 2323–2331 (ISSN 0012-186X et 1432-0428, DOI 10.1007/s00125-007-0794-x, lire en ligne, consulté le )
- (en) Paul S Ciechanowski, Wayne J Katon, Joan E Russo et Irl B Hirsch, « The relationship of depressive symptoms to symptom reporting, self-care and glucose control in diabetes », General Hospital Psychiatry, vol. 25, no 4, , p. 246–252 (DOI 10.1016/S0163-8343(03)00055-0, lire en ligne, consulté le )
- (en) « U.K. prospective diabetes study 16. Overview of 6 years' therapy of type II diabetes: a progressive disease. U.K. Prospective Diabetes Study Group », Diabetes, vol. 44, no 11, , p. 1249–1258 (ISSN 0012-1797 et 0012-1797, DOI 10.2337/diabetes.44.11.1249, lire en ligne, consulté le )
- (en) A.S. Rudenski, D.R. Matthews, J.C. Levy et R.C. Turner, « Understanding “insulin resistance”: Both glucose resistance and insulin resistance are required to model human diabetes », Metabolism, vol. 40, no 9, , p. 908–917 (DOI 10.1016/0026-0495(91)90065-5, lire en ligne, consulté le )
- (en) Run Yu, Nicholas N. Nissen, Andrew Hendifar et Laura Tang, « A Clinicopathological Study of Malignant Insulinoma in a Contemporary Series », Pancreas, vol. 46, no 1, , p. 48–56 (ISSN 0885-3177, DOI 10.1097/MPA.0000000000000718, lire en ligne, consulté le )
- (en) Shari Bolen, Leonard Feldman, Jason Vassy et Lisa Wilson, « Systematic Review: Comparative Effectiveness and Safety of Oral Medications for Type 2 Diabetes Mellitus », Annals of Internal Medicine, vol. 147, no 6, , p. 386 (ISSN 0003-4819, DOI 10.7326/0003-4819-147-6-200709180-00178, lire en ligne, consulté le )
- (en) S. E. Inzucchi, R. M. Bergenstal, J. B. Buse et M. Diamant, « Management of hyperglycaemia in type 2 diabetes: a patient-centered approach. Position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD) », Diabetologia, vol. 55, no 6, , p. 1577–1596 (ISSN 0012-186X et 1432-0428, DOI 10.1007/s00125-012-2534-0, lire en ligne, consulté le )
- (en) Nicholas B. Whitticar, Elisha W. Strahler, Parthiban Rajan et Savas Kaya, « An Automated Perifusion System for Modifying Cell Culture Conditions over Time », Biological Procedures Online, vol. 18, no 1, , p. 19 (ISSN 1480-9222 et 1480-9222, PMID 27895534, PMCID PMC5117600, DOI 10.1186/s12575-016-0049-7, lire en ligne, consulté le )
- (en) Z. Wang et H. Gleichmann, « GLUT2 in pancreatic islets: crucial target molecule in diabetes induced with multiple low doses of streptozotocin in mice », Diabetes, vol. 47, no 1, , p. 50–56 (ISSN 0012-1797 et 0012-1797, DOI 10.2337/diabetes.47.1.50, lire en ligne, consulté le )
- (en) I.G. Danilova, P.A. Sarapultsev, S.U. Medvedeva et I.F. Gette, « Morphological Restructuring of Myocardium During the Early Phase of Experimental Diabetes Mellitus: Restructuring of Myocardium in Diabetes », The Anatomical Record, vol. 298, no 2, , p. 396–407 (DOI 10.1002/ar.23052, lire en ligne, consulté le )
- Aileen JF King, « The use of animal models in diabetes research: Animal models of diabetes », British Journal of Pharmacology, vol. 166, no 3, , p. 877–894 (DOI 10.1111/j.1476-5381.2012.01911.x, lire en ligne, consulté le )