Système rénine-angiotensine-aldostérone
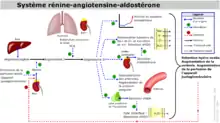
Le système rénine-angiotensine-aldostérone (SRAA ou RAAS pour les anglophones) est chez les mammifères l'un des systèmes de régulation les plus importants des fonctions autonomes, cardiovasculaires et pulmonaires[1] ; il s'agit d'une cascade de régulation endocrinienne et enzymatique. C'est un système hormonal organisé autour du rein, qui permet notamment de préserver l'homéostasie hydrosodée (l'équilibre entre les ions Na+ et l'eau).
Métabolisme
Voie dépendante de l'enzyme de conversion de l'angiotensine
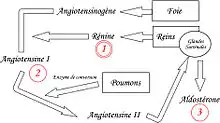
L'angiotensinogène, protéine inactive produite par le foie, circule dans le sang. C'est le précurseur des peptides actifs angiotensine I et II, et le seul substrat de la rénine.
En cas de baisse de la pression dans l'artère rénale, la rénine (enzyme parfois considérée comme une hormone) est sécrétée au niveau du rein par des cellules myoépithéliales différenciées de l’artériole afférente de l’appareil juxtaglomérulaire[2]. Il existe également d'autres stimuli favorisant la sécrétion de rénine : baisse de la natrémie au niveau du tube contourné distal, ß-agonistes, hyperkaliémie, PGI2 et stimulation des cellules juxta-glomérulaires par le système nerveux sympathique[3])
L'angiotensinogène, sécrété par le foie, est clivée par la rénine et forme un décapeptide appelé « angiotensine I », inactif.
L'angiotensine I sera ensuite principalement transformée en angiotensine II par une carboxypeptidase, l'enzyme de conversion de l'angiotensine (ECA, ou ACE pour les Anglo-Saxons). Cette enzyme, sécrétée par le foie, agit au niveau pulmonaire.
Voie non dépendante de l'enzyme de conversion de l'angiotensine
Il existe des enzymes (cathepsine G et t-PA) permettant de passer directement de l’angiotensinogène à l’angiotensine II, et d’autres enzymes (chymases, CAGE) jouant le même rôle que l’ECA (transformer l'angiotensine I en angiotensine II). Ces voies « non-ECA » représentent jusqu’à 40 % de la synthèse de l’angiotensine II[4] - [5] - [6].
Rôles
Angiotensine II
L'angiotensine agit en se fixant sur ses récepteurs transmembranaires. Il existe deux types de récepteurs, AT1 (majoritaire) et AT2, qui ont des rôles antagonistes[7].
Le récepteur AT2 est le plus rare, et n'est pas inhibé par les traitements par antagoniste de récepteur de l'angiotensine II. Il est responsable de vasodilatation, inhibition de croissance cellulaire et apoptose.
Via le récepteur AT1 (récepteur couplé à une protéine G), l'angiotensine II favorise l'élevation de la pression artérielle par différents mécanismes :
- stimulation de la vasoconstriction des artérioles (directe et indirecte, via le relarguage de noradrénaline), provoquant une augmentation des résistances périphériques et le maintien de la filtration glomérulaire (par vasoconstriction de l'artériole efférente) ;
- hyperplasie et hypertrophie vasculaire (initiés par les mêmes voies intracellulaires de phosphorylation de Tyrosine que ceux utilisés par les cytokines)[8] ;
- stimulation de la réabsorption tubulaire de sodium (Na+) ;
- sécrétion d'aldostérone par la partie glomérulée du cortex surrénalien (action sur la pompe sodium-potassium, entraînant une réabsorption de sodium (3 Na+) et d'eau contre des ions potassium (2 K+) ;
- stimulation de la sécrétion de vasopressine (encore appelée hormone antidiurétique ou ADH) qui limite la perte d'eau dans les urines, ce mécanisme ayant lieu au niveau du tube contourné distal et du tube collecteur[3] - [9] ;
- stimulation de la sensation de soif, entrainant une plus grande absorption d'eau qui mécaniquement augmentera le volume sanguin et donc la pression artérielle.
Autres angiotensines
L’angiotensine I peut être transformée en angiotensine-(1-7) par des endopeptidases neutres (néprilysine, ECA2 exprimée au niveau rénal et cardiaque). L’angiotensine-(1-7), comme l’angiotensine IV (fragment de protéolyse de l’angiotensine II), augmente la synthèse et la sécrétion de prostaglandines relaxantes, potentialise l’action de la bradykinine[10].
Au total, le couple ECA2/angiotensine-(1-7) apparaît comme un élément essentiel de la contre-régulation des actions du couple ECA/angiotensine II[11].
Effets annexes de l'enzyme de conversion de l'angiotensine
En parallèle de son action sur l’angiotensine I, l’ECA a également un rôle au niveau du système kinine-kallicréine. En effet, dans ce système, les kiniogènes de haut poids moléculaires sont dégradées en bradykinine par la kallicréine plasmatique. Les bradykinines sont vasodilatatrices de par leur action sur la libération de prostaglandines (PGE2 et PGI2), et leur fixation sur le récepteur B2. Elles sont dégradées en peptides inactifs par deux kininases principales, dont l’enzyme de conversion de l'angiotensine (ou kininase II)[3].
Au total, l'enzyme de conversion de l'angiotensine stimule la vasoconstriction en générant l'angiotensine II (peptide vasoconstricteur) et en inactivant la bradykinine (peptide vasodilatateur). Ce mécanisme explique en partie[12]les effets spécifiques des inhibiteurs de l'enzyme de conversion, toux sèche et irritative (5 à 35 % des patients traités[13]) et œdème de Quincke (0,1 à 0,5 %[14])
Contrôle du système rénine-angiotensine-aldostérone (SRAA)
Le blocage du système rénine-angiotensine-aldostérone (SRAA), que ce soit au niveau systémique ou au niveau tissulaire local, joue un rôle protecteur contre plusieurs pathologies cardiaques ou rénales[15]. Inhiber le système rénine-angiotensine-aldostérone (SRAA) est devenu l'un des meilleurs moyens de soigner l'hypertension artérielle[15] de la décompensation cardiaque[16], de la protection postinfarctus[17] - [18] et de la néphropathie avec microalbuminurie ou protéinurie, notamment la néphropathie diabétique[19]. C'est aussi une piste explorée contre le diabète[20].
Régulation du système rénine-angiotensine
Régulation physiologique
Pour stopper les effets de l'angiotensine II, il faut stopper sa sécrétion.
Ceci se fait via deux rétrocontrôles négatifs :
- l'angiotensine II a un effet inhibiteur sur la sécrétion de rénine, c’est-à-dire que, plus la concentration en angiotensine II augmente, plus la concentration en rénine diminue. Donc il y a de moins en moins d'angiotensine II formée.
- le second rétrocontrôle négatif est dû à l'action de l'aldostérone, notamment par la régulation de l'expression du canal épithélial à sodium ou ENaC et de la pompe sodium-potassium dans le tube distal du rein entraînant une rétention de sodium et d'eau, donc l’augmentation de la pression artérielle. L'augmentation de la pression artérielle au niveau de l'appareil juxtaglomérulaire du rein va fortement inhiber la sécrétion de rénine.
Médicaments régulateurs
De nombreux médicaments antihypertenseurs bloquent cette cascade de réactions à différents niveaux pour faire baisser la pression artérielle :
- IEC : Inhibiteurs de l'enzyme de conversion de l'angiotensine (captopril, énalapril, ramipril, etc.).
- ARA2 : Antagonistes du Récepteur de l'Angiotensine II, ou bloqueurs du récepteur AT1 (valsartan, losartan, telmisartan, candésartan, etc.).
- Diurétiques
- Inhibiteur de la rénine (depuis [21]) : aliskiren
Les IEC et ARA2 cassent la boucle de rétrocontrôle négatif de l’angiotensine II sur la rénine et entraînent donc une augmentation de l’activité rénine plasmatique (ARP), de la concentration plasmatique en angiotensine I (et également d’angiotensine II sous ARA2[22]).
Sous ARA2, l’expression des récepteurs AT2 et des récepteurs de la bradykinine est également augmentée, à cause de cette augmentation de l’activité rénine plasmatique[23].
Avec les inhibiteurs de la rénine, la production de rénine est augmentée (via la boucle de rétrocontrôle), mais sa capacité pour former l’angiotensine I (i.e. son activité rénine plasmatique) est réduite de 80 %[24] - [25].
Nicotine ?
On sait depuis au moins les années 1970 ans que l'inhalation de nicotine augmente la pression sanguine (systolique et diastolique)[26] - [27] - [28] conjointement à une activité augmentée de l'ACE[27],
La nicotine semble fortement agir sur le système rénine-angiotensine (RAS) en en modifiant l'homéostasie ; en régulant positivement l'axe des récepteurs de l'enzyme de conversion de l'angiotensine (ACE)/angiotensine (ANG)-II/ANG II de type 1 et en régulant à la baisse l'ACE2/ANG compensatoire ACE2/ANG-(1–7)/Axe des récepteurs Mas, contribuant au développement de la CVPD[29].
Voir aussi
Articles connexes
Bibliographie
- Atkinson, J., Dupuis, F., & Chillon, J. M. (2007, May). Systeme renine-angiotensine-aldosterone: vieux systeme mais piste strategique de regulation de la circulation cerebrale. In Annales pharmaceutiques françaises (Vol. 65, No. 3, pp. 195-202). Elsevier Masson (résumé).
- Damy, T., Guellich, A., Vermes, E., Deswarte, G., & Hittinger, L. (2007) Physiologie et physiopathologie du système rénine-angiotensine-aldostérone. mt cardio, 3(4), 257-262 (résumé).
- De Groote, P. rénine-angiotensine-aldostérone: comment associer les médicaments? .
- Dupuis, F. (2005). Implication du système rénine angiotensine aldostérone dans les altérations de la circulation cérébrale au cours de l'hypertension artérielle chronique (Doctoral dissertation, Université Henri Poincaré-Nancy 1).
- Gomes-Feirera, S., Champ-Rigot, L., Benard, L., Scanu, P., Hurpe, J. M., Samuel, J. L., ... & Milliez, P. (2008). Existe-t-il un lien entre la fibrillation auriculaire et le système rénine-angiotensine-aldostérone?. MT Cardio, 4(3), 194-200 (résumé).
- Legrand, D., Krzesinski, J. M., & Scheen, A. (2008). Quelle place pour une double ou triple inhibition du système rénine-angiotensine-aldostérone?. Revue médicale suisse, 4(168), 1792-7.
- Morel, B. (2007). Le système rénine-angiotensine-aldostérone chez le chien sain et le chien insuffisant cardiaque (Doctoral dissertation)
- Ton A.T (2019) Le rôle du système rénine-angiotensine et de la différence liée au sexe dans la fibrillation auriculaire chez la souris (résumé).
Notes et références
- Mckinley, Anatomie et physiologie, Canada, chenelière éducation,
- L. Amara, A.-P. Gimenez-Roqueplo, P. Rossignol et P.-F. Plouin, « Tumeurs à rénine », EMC-Endocrinologie, vol. 2, no 2, , p. 121-127 (résumé)
- Rang H.P., M.M. Dale et al. Rang and Dale's Pharmacology (2007) pp. 302-303
- Norman K. Hollenberg, Naomi D.L. Fisher et Deborah A. Price, « Pathways for angiotensin II generation in intact human tissue : evidence from comparative pharmacological interruption of the renin system », Hypertension, vol. 32, no 3, , p. 387-92 (lire en ligne)
- H.L. Jackman, M.G. Massad, M. Sekosan et al., « Angiotensin 1-9 and 1-7 release in human heart:role of cathepsin A », Hypertension, vol. 39, no 5, , p. 976-81 (lire en ligne)
- Leckie B.J. Targeting the Renin-angiotensin System: What's new ? Curr Med Chem - Cardiovascular & Hematological Agents, 2005, 3, 23-32
- (en) Peterson R.C. et al. « Angiotensin II Receptor Blockers in Heart Failure: Role of the RAAS in the Pathophysiology of Heart Failure » sur www.medscape.com
- (en) Marrero et al. Nature 1995;375:247-250
- Il est à noter que la réabsorption d'eau étant supérieure à la réabsorption de sel par cette double action de l'aldostérone (pompe Na/K et ADH), on trouve ici l'explication de l'hyponatrémie de dilution lors des hyperhydratations intracellulaires !
- « unilim.fr/theses/2005/sante/20… »(Archive.org • Wikiwix • Archive.is • Google • Que faire ?).
- Campell DJ. Renin inhibitors – mechanisms of action. Aust Prescr 2009;32:132-5
- Note : Leur mécanisme n’est pas encore totalement élucidé, mais la majoration du taux de bradykinine et de substance P, normalement dégradés par l’ECA, et celui de prostaglandines PGE2 et PGI2 (stimulées par la bradykinine et inhibées par indométacine) en sont en partie responsables. Parmi les autres mécanismes évoqués, citons la stimulation des fibres vagales, le polymorphisme du gène promoteur du récepteur B2 de la bradykinine menant à une augmentation de la densité de ces récepteurs selon Mukae S.)
- Dicpinigaitis PV. Angiotensin-Converting Enzyme Inhibitor-Induced Cough ACCP Evidence-Based Clinical Practice Guidelines
- selon le Martindale (données pharmacologiques anglo-saxonnes)
- White WB. Angiotensin-converting enzyme inhibitors in the treatment of hypertension : An update. J Clin Hypertens 2007;9:876-82
- Jorde UP. Suppression of the renin-angiotensin-aldosterone system in chronic heart failure : Choice of agents and clinical impact. Cardiol Rev 2006;14:81-7
- Cohn JN. Reducing cardiovascular risk by blockade of the renin-angiotensin- aldosterone system. Adv Ther 2007;24:1290-304
- Schmieder RE, Hilgers KF, Schlaich MP, Schmidt BM. Renin-angiotensin system and cardiovascular risk. Lancet 2007;369:1208-19
- Gurley SB, Coffman TM. The renin-angiotensin system and diabetic nephropathy. Semin Nephrol 2007;27: 144-52
- Scheen AJ. Renin-angiotensin system inhibition prevents type 2 diabetes mellitus. Part 1. A meta-analysis of randomised clinical trials. Diabetes Metab 2004;30: 487-96
- (en) « First Hypertension Drug to Inhibit Kidney Enzyme Approved »(Archive.org • Wikiwix • Archive.is • Google • Que faire ?), CBC, (consulté le )
- Shibasaki Y, Mori Y, Tsutumi Y, Masaki H, Sakamoto K, Murasawa S, Maruyama K, Moriguchi Y, Tanaka Y, Iwasaka T, Inada M et Matsubara H. (1999) Differential kinetics of circulating angiotensin IV and II after treatment with angiotensin II type 1 receptor antagonist and their plasma levels in patients with chronic renal failure. Clin Nephrol, 51(2), 83-91.
- Tschope C, Spillmann F, Altmann C, Koch M, Westermann D, Dhayat N, Dhayat S, Bascands JL, Gera L, Hoffmann S, Schultheiss HP et Walther T. (2004) The bradykinin B1 receptor contributes to the cardioprotective effects of AT1 blockade after experimental myocardial infarction. Cardiovasc Res, 61(3), 559-569.
- Vaidyanathan S. et al. Clinical pharmacokinetics and pharmacodynamics of aliskiren. Clin Pharmacokinet. 2008 ;47(8) :515-31
- Brookes L. New Data on the Oral Renin Inhibitor Aliskiren in Patients with Hypertension.
- (en) Georgio U. Cellina, A.John Honour et William A. Littler, « Direct arterial pressure, heart rate, and electrocardiogram during cigarette smoking in unrestricted patients », American Heart Journal, vol. 89, no 1, , p. 18–25 (DOI 10.1016/0002-8703(75)90004-6, lire en ligne, consulté le )
- García Calzado MC, García Rojas JF, Mangas Rojas A, Millán J. Tobacco and arterial pressure (II.). The acute effects on the angiotensin-converting enzyme [in Spanish]. An Med Interna 7: 392–395, 1990
- (en) Antonella Groppelli, Dante M. A Giorgi, Stefano Omboni et Gianfranco Parati, « Persistent blood pressure increase induced by heavy smoking: », Journal of Hypertension, vol. 10, no 5, , p. 495–499 (ISSN 0263-6352, DOI 10.1097/00004872-199205000-00014, lire en ligne, consulté le )
- (en) Joshua M. Oakes, Robert M. Fuchs, Jason D. Gardner et Eric Lazartigues, « Nicotine and the renin-angiotensin system », American Journal of Physiology-Regulatory, Integrative and Comparative Physiology, vol. 315, no 5, , R895–R906 (ISSN 0363-6119 et 1522-1490, PMID 30088946, PMCID PMC6295500, DOI 10.1152/ajpregu.00099.2018, lire en ligne, consulté le )