Synthèse de complexes du nickel
Cet article traite la synthèse de trois différents complexes du nickel dont le nombre d’atomes coordonnés varie de 4 à 6, ainsi que leurs propriétés optiques et magnétiques.
Principe
Beaucoup de composés organiques, qui contiennent 2 atomes ou plus capables de coordonner en ions métalliques, agissent comme des ligands. Avec des ions métalliques bivalents, il est souvent facile de les isoler de la solution complexe aqueuse n’ayant pas de charge globale (les charges négatives des ligands équivalents les charges positives de l’ion métallique). Ainsi, si le composé organique a une charge négative (salicylaldéhyde), le complexe qui contient deux ligands organiques et des ions bivalents métalliques a une solubilité faible dans l’eau. Les complexes préparés dans cette expérience sont les dérivés nickel (II) des phénols substitués.
Par un choix approprié du composé organique, il est possible de préparer des complexes métalliques dans lesquels le nombre d’atomes coordonnés à l’ion central varie (ici de 4 à 6). Dans cette expérience, les complexes synthétisés sont :
- le bis (salicylaldéhydato) nickel (II) dihydraté : complexe I ;
- le bis (salicylidèneiminato) nickel (II) : complexe II ;
- le bis (N-s butylsalicylidèneiminato) nickel (II) : complexe III.
Le complexe I a une structure contenant 6 atomes d’oxygène coordonnés des trois types différents. Les composés imines ou bases de Schiff, ressortent quand le salicylaldéhyde est traité avec de l’ammoniac ou certaines amines (méthylamine ou éthylamine) peuvent former des complexes similaires bien qu’il n’y ait plus que 4 atomes coordonnés (2 azote et 2 oxygène). Les 4 atomes coordonnés sont tous en plan comme illustré dans le schéma de synthèse. Bien qu’il ne soit pas immédiatement évident, la distance entre le groupe N-R et l’oxygène voisin est très faible. Si R est grand, il n’est pas forcément possible d’avoir les atomes coordonnés dans le plan. Pour le complexe III, la structure aura donc un arrangement tétraédrique.
La couleur des complexes formés est différente bien que pour les complexes II et III les atomes coordonnés au métal sont les mêmes. La couleur est due à l’absorption de la lumière favorisant les électrons, et spécialement les électrons d aux niveaux d’énergie les plus élevées associées au nickel. Les niveaux et par conséquent les couleurs sont affectés par le changement de la stéréochimie des complexes.
Le complexe I est paramagnétique, les électrons sont attirés par un champ magnétique. Le complexe II est diamagnétique, les électrons sont repoussés par un champ magnétique. Le complexe III a des propriétés physiques intermédiaires.
Nous pourrons déterminer la teneur en nickel des complexes formés par dosage gravimétrique. Nous ne pourrons cependant effectuer cette analyse que sur le complexe I et III car le complexe II reste insoluble. Le dosage gravimétrique consiste à premièrement solubiliser les ions Ni2+. Nous les rendrons ensuite insolubles en les faisant complexer avec la diméthylglyoxime (DMG). Grâce à un excès d’ammoniaque, le complexe va précipiter. Nous n’aurons plus qu’à filtrer la solution et peser le solide obtenu pour pouvoir calculer la concentration en ion Ni2+. L’ammoniaque est utilisé car le précipité devient insoluble dans les solutions ammoniacales diluées.
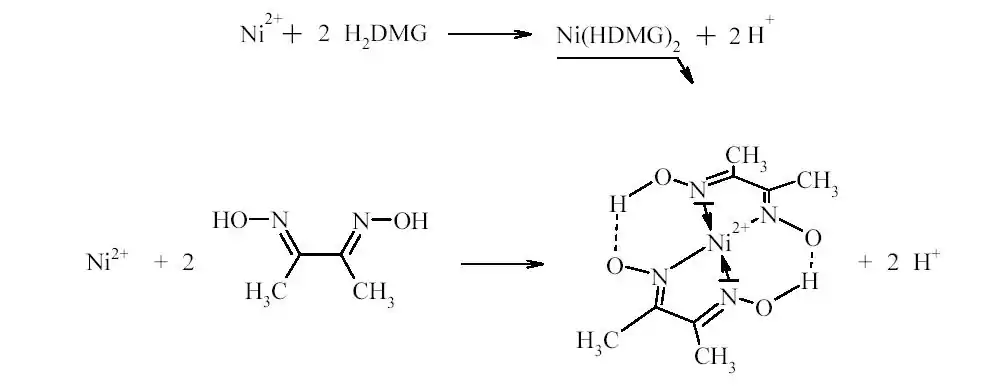
Schéma de la synthèse
Formation du complexe I :
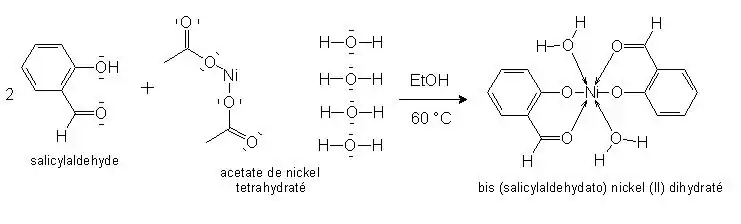
La réaction mise en œuvre ici est une réaction de complexation. Pour cette complexation, les ligands seront :
- le salicylaldéhyde qui sera ici un ligand bidentate car il se fixe à l’ion métallique par deux atomes en même temps ;
- l’eau qui est un ligand monodentate.
La purification du complexe se fera par une simple filtration puis un lavage du fait de sa faible solubilité. La caractérisation du complexe pourra se faire par spectroscopie infrarouge (IR) car nous avons pu trouver un spectre de référence. L’analyse CHNS-O permettra de déterminer le pourcentage d’atomes de carbone, d'hydrogène, d'azote et de soufre contenu dans la molécule.
Formation du complexe II à partir du complexe I obtenu précédemment :
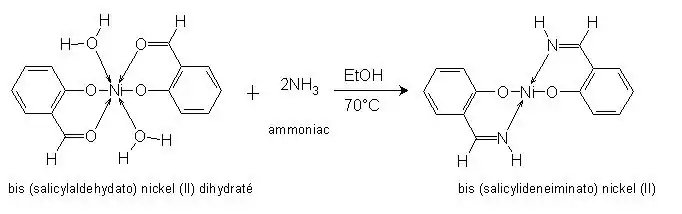
Pour former ce complexe, nous procèderons à une substitution de l’atome d’oxygène par un atome d’azote qui entraine une déshydratation du complexe. Le ligand est à nouveau bidentate. La purification du complexe se fera par une simple filtration puis un lavage du fait de sa faible solubilité. La caractérisation du complexe ne pourra pas se faire par spectroscopie infrarouge car nous n’avons pas pu trouver un spectre de référence. L’analyse CHNS-O permettra de déterminer le pourcentage d’atomes de C, H, N, S contenu dans la molécule.
Formation du complexe III à partir du complexe I obtenu précédemment :
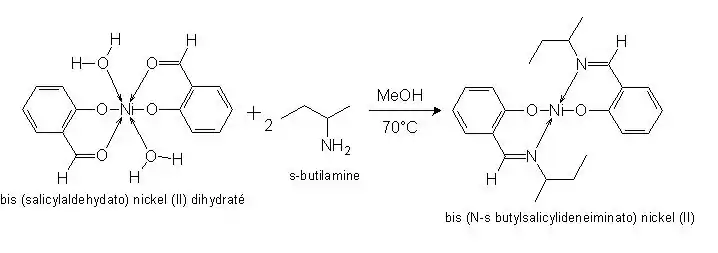
Pour former ce complexe, nous procèderons à une substitution de l’atome d’hydrogène par la chaîne carbonée de la sec-butylamine. Le ligand est à nouveau bidentate. La purification du complexe se fera par une simple filtration puis un lavage du fait de sa faible solubilité. La caractérisation du complexe ne pourra pas se faire par spectroscopie infrarouge car nous n’avons pas pu trouver un spectre de référence. L’analyse CHNS-O permettra de déterminer le pourcentage d’atomes de C, H, N, S contenu dans la molécule.
Mode opératoire
- Synthèse du bis (salicylaldéhydato) nickel (II) dihydrate (I)
Dissoudre 15 g de fine poudre d’acétate de nickel tétrahydraté dans 250 ml d’eau, dans un ballon de 1 L. Ajouter 250 ml d’éthanol et doucement 12,6 ml de salicylaldéhyde en mélangeant vigoureusement. Chauffer la solution au bain-marie et a reflux pendant 30 min en agitant fréquemment pour homogénéiser la réaction. Laisser refroidir (pendant au moins 1 h), filtrer le précipité cristallin et laver à l’eau, à l’éthanol puis avec un minimum d’éther. Sécher le produit à l’étuve (70 à 100 °C), on obtient un solide de couleur vert clair.
L’acétate de nickel tétrahydraté peut être difficile à dissoudre dans l’eau, mais en chauffant légèrement il se dissout en 20 min. Il faut faire attention à ne pas chauffer trop rapidement sinon il se forme un précipité insoluble qu’il faut filtrer avant d’ajouter de l’éthanol.
- Synthèse du bis (salicylideineiminato) nickel (II)
!!!!!Manipuler impérativement sous la hotte : présence d’ammoniac !
Chauffer au bain-marie et à reflux dans un ballon de 250 ml une suspension de 3,4 g de bis (salicylaldéhydato)-nickel (II) di hydraté dans 20 ml d’éthanol et 20 ml d’une solution d’ammoniac à 35 %. Chauffer jusqu'à ce que la solution reflue. Agiter doucement le ballon à intervalles réguliers jusqu'à ce que la réaction soit complète (30 min). Refroidir, filtrer le précipité cristallin et laver à l’éthanol puis à l’éther. On obtient une solution de couleur rouge.
- Synthèse du bis (N-s-butylsalicylideneiminato) nickel (II)
Chauffer au bain marie et à reflux dans un ballon de 250 ml une suspension de 3,4 g de bis (salicylaldéhydato) nickel (II) di hydraté, dans 25 ml d’éthanol et 2,25 ml de sec-butylamine. Chauffer jusqu'à ce que la réaction soit complète (30 min). Filtrer la solution chaude et laisser refroidir doucement, on obtient une solution verte et il faut laisser reposer pendant au moins 1 h 30 pour voir apparaître des cristaux vert au fond du ballon. Filtrer le précipité cristallin (vert) et laver avec un minimum de méthanol froid. Sécher le produit à l’air puis dans l’étuve (40 à 50 °C) puis pas plus de 70 °C sinon le produit est détérioré.
- Dosage gravimétrique des ions nickel
Avant toute manipulation, laver et faire sécher à l’étuve pendant au moins 3 h, 2 gooch ; une fois qu’ils sont bien secs, noter leur masse à vide. (1 : 50,890 g ; 2 : 49,421 g) Réaliser 2 fois cette manipulation en parallèle. Peser et placer dans un bécher 0,25 g du complexe à doser. Ajouter 10 ml de HCl concentré et diluer avec environ 100 ml d’eau. Chauffer à 70 °C. Ajouter 25 ml d’une solution de diméthylglyoxime dans l’éthanol à 1 % massique. Ajouter immédiatement après la DMG de l’ammoniac concentré goutte à goutte jusqu'à obtenir un pH=9. Laisser reposer 30 à 40 min tout en agitant la solution (rouge). Sortir les gooch de l’étuve et placer-les dans un dessiccateur (pour éviter qu’il prenne l’humidité). Pendant que les solutions reposent, vérifier en prélevant quelques gouttes du liquide surnageant et ajouter de la DMG s’il y a formation d’un précipité rouge alors la réaction n’est pas complète et il faut rajouter un peu de DMG. Une fois les solutions refroidies ainsi que les gooch, filtrer sous vide les solutions à travers les gooch, puis laver avec de l’eau et faire sécher à l’étuve (110 à 120 °C). Peser les gooch puis les remettre à l’étuve et re-peser jusqu'à obtenir deux fois la même masse (on obtient un solide rouge).